06
2022.04
三代定序策略與案例 │ 邁向大型族群研究
原創文章 引用請註明出處
? 為什麼西方人皮膚是白的?
? 為什麼人口平均身高最高的國家幾乎都在歐洲?
? 為什麼心臟病、糖尿病與氣喘等常見的慢性病,發生在非裔美國人的比率高於非拉丁裔的白人?
… (以下省略一百個問題,絕對是因為篇幅問題,不是小編偷懶喔)
最後要問:
為什麼每個族群看起來那麼不同卻都使用同一個參考基因體呢?
最後一個問題也是現在各國積極推動大型族群研究的原因
--- 建立該國的人群資料庫
背景及挑戰:
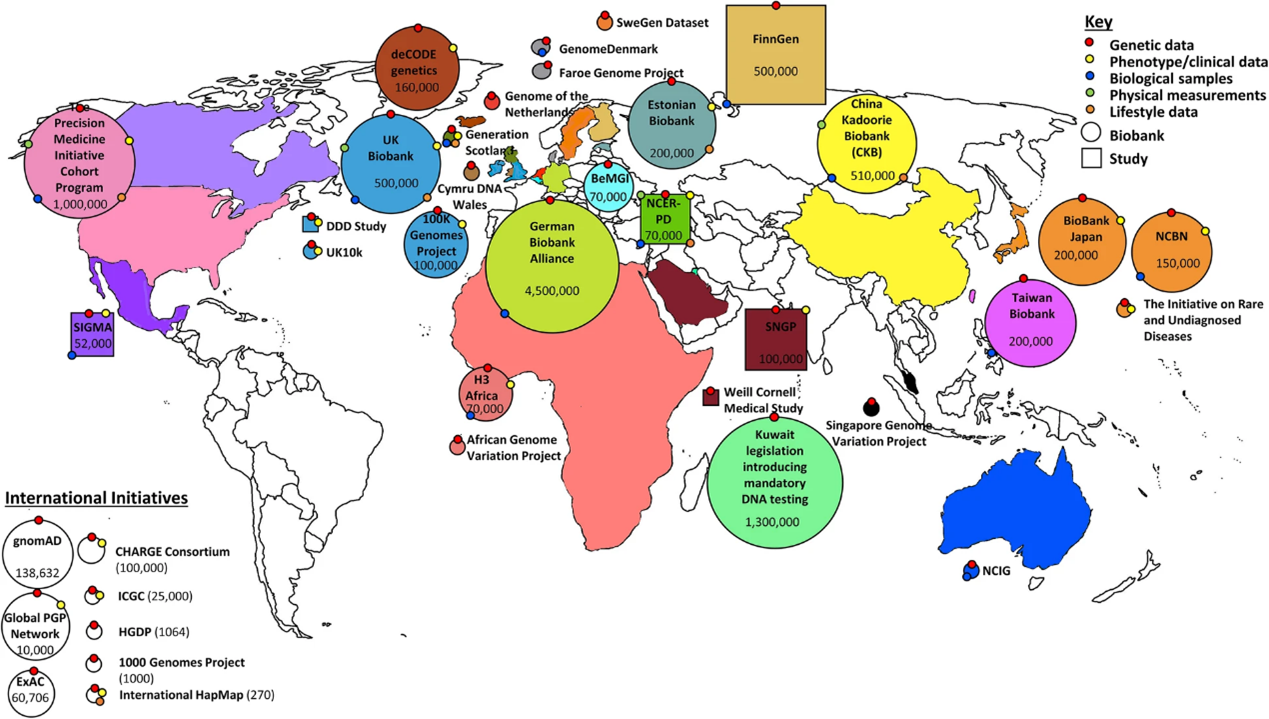
事實上,早在 1948 年開始,世界各地陸續開始成立國家型人體生物資料庫 (圖一),包含美國、英國、中國、日本、韓國等國家,期望這些資料庫能發掘族群特有的遺傳變異、對於複雜的遺傳疾病能更好理解以及快速發現,從而實現精準化治療。
以前的族群研究,包括全基因體關聯研究 (GWAS),並未能詳盡地描述人類特徵和遺傳疾病的相關性 [2]。關於這種“遺傳性缺失”的來源有很多猜測,通常指向結構變異 (SV) 和罕見變異。
在人類基因體中,SVs 所影響的核苷酸總數比單核苷酸變異 (SNV) 數量多得多。但直到目前為止,此類族群研究仍主要依賴於二代定序技術完成,該技術產生的讀長範圍為 25 bp 至 600 bp。
然而,二代定序的短讀長在重複區域方面遭遇到嚴重限制。DNA 重複序列作為基因體單元促進了 SV 的形成,同時也由於序列無法準確對齊而阻礙了 SV 的發現。即使在非重複區域中,二代定序由於技術限制也容易遺漏插入等變異(尤其是長度超過定序讀長的等位基因) 或其他修飾(例如甲基化)。隨著三代定序的成熟,越來越多研究開始將三代定序作為基因計畫的技術平台。
使用三代定序之大型研究:
瓶中基因體 (GIAB) 和人類基因體結構變異 (HGSV) 聯盟結合了多種技術來全面表徵人類基因體的結構變異,不約而同表示,三代定序在結構變異識別的能力已遠優於二代序和其他方法(例如 arrays)[3] [4] 。這些研究強調,通過三代定序可以發現很大一部分隱藏的變異。
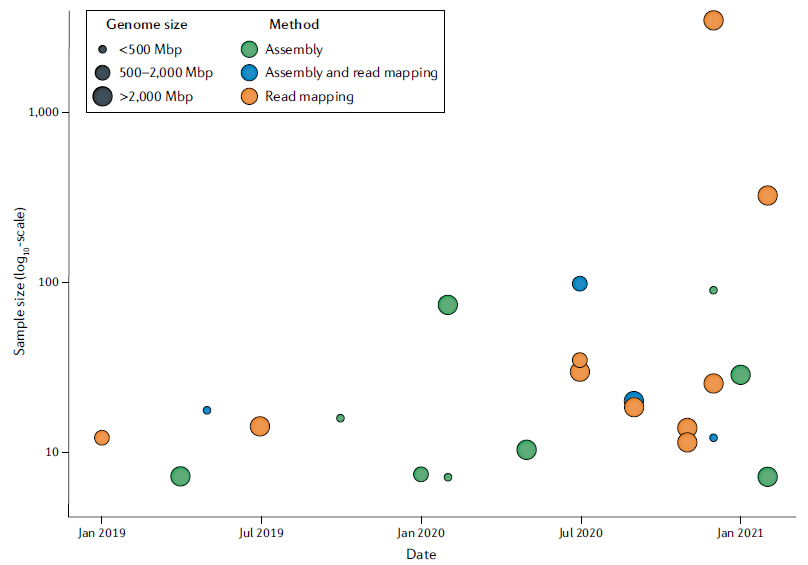
已經有越來越多的大型研究納入三代定序作為發掘 SVs 的工具,從 2019 年到 2021 年使用三代定序且超過 5 個樣本的研究統計如圖二; 詳細列表於表一。
定序策略:
在大規模人群定序項目開始時,需要考慮多種具有特定預算要求的常用策略(圖三)。
下列討論三種主要策略,以達成不同的規模和預算,但須注意其對檢測遺傳變異的辨別率會產生影響。
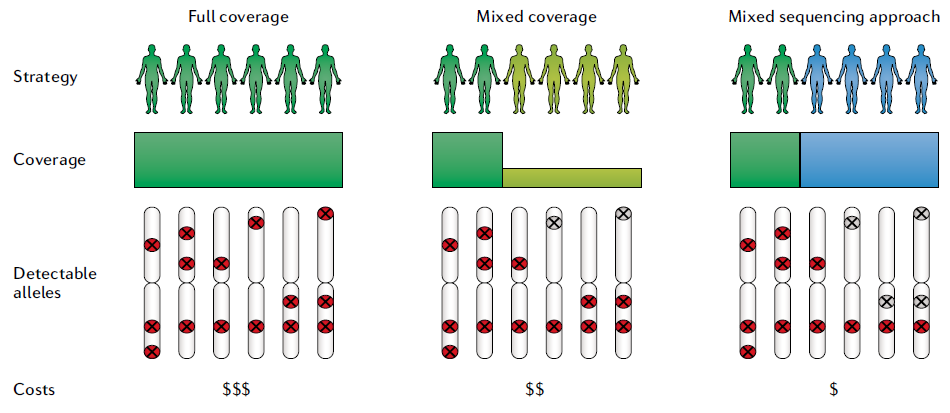
(左) 全覆蓋策略(Full coverage):
所有樣本都以中 ( >12x 定序深度) 到高 (>40x 定序深度) 的三代定序深度進行定序。儘管其成本為這三種方法中最高,但其優勢在於全面性、高水平的分辨率、研究設計的簡單性和相對簡單的計算工作流程。
假設: 20x 的覆蓋度對 2,500 人進行定序,需要 150 Tb的數據量。
(中) 混合覆蓋策略 (Mixed coverage) :
代表樣本 (如: 亞種) 以中到高覆蓋度 (30x 定序深度) 進行三代定序,其餘樣本使用低覆蓋度 (>5x 定序深度) 進行三代定序(類似於最初的 1000 Genomes 項目)。混合覆蓋策略雖然比全覆蓋策略便宜,但仍然可以實現較高的整體檢測靈敏度,因此特別適用於人數眾多或預算有限的研究。但是,分析具有較高挑戰,特別是在跨多個樣本實現準確基因型預測、或是區分體細胞與雜合種系變異等。此外,混合覆蓋策略偏向發現常見等位基因,許多稀有等位基因可能會被遺漏。
假設: 30x 的覆蓋度對 200 人定序,其餘個體以 8x 覆蓋度進行定序,僅需要 73 Tb 的數據量,是全覆蓋策略的一半。
(右) 混和定序策略 (mixed sequencing approach)
一部分樣本 (10~20%) 以三代定序進行中到高覆蓋度定序,其餘樣本使用二代定序。採用哪種方法的決定將影響檢測人群中常見(紅色符號)或罕見(灰色符號)變異的能力。該決定還取決於可用預算、現有數據和樣本 DNA 可用性。混和定序策略的原理類似於混合覆蓋策略,可有效闡明易患癌症的種系 SV,其中二代定序用於驗證 SV。有些研究團隊使用 SVCollector [6] 自動選擇樣本進行三代定序,其識別的 SV 以現有的二代定序數據集中對 SV 的斷點坐標進行基因分型(例如,插入)。通過這種方式,可以獲得已識別變體的穩健等位基因頻率,但依然可能會遺漏其他樣本中包含的稀有變體。
假設: 30x 的覆蓋度對 200 人定序,其餘個體使用二代定序數據,僅需要 18 Tb 的數據量,是最經濟實惠的選項。
最新案例:

迄今為止,最大規模且以人類為中心的三代定序研究於 2021 發表於 Nature Genetics [7],本篇研究利用 Nanopore 平台檢測 3,622 位冰島人基因體多樣性,發現每位冰島人平均擁有 22,636 個結構變異,包含了 13,353 個插入及 9,474 個缺失變異。這些變異中,含有 133,886 個可信的等位基因結構變異。接著,在 166,281 位冰島人中,驗證這些變異與疾病之間的關係。研究結果發現,擁有 PCSK9 基因第一個外顯子缺失者,其低密度脂蛋白的濃度較一般人平均濃度低。而另一個基因 ACAN,有長度為 57 bp 的多個重複序列,包含了 11 個等位基因型,其重複次數與個體身高有著顯著相關。這篇研究證實了,三代定序長讀長的特點,可以有效的在族群間利用全基因體定序找到有意義的結構變異,甚至解密這些結構變異對於人類表型的影響。[8]
當然,除了本篇的冰島人基因體定序計畫,還有阿布達比的 100,000 人基因定序計畫、NIH All of Us 研究計劃和 NIH 阿茲海默症和相關癡呆症中心 (CARD) 在美國和中國、阿布扎比和卡塔爾的類似研究,都可以從中看出三代定序的應用層面逐漸擴大與普及。
除了人類層面的研究之外,三代定序已在族群規模上應用,以發現與作物 [9] [10]、果蠅 [11] 和鳴禽 [12] 的表型相關的結構變異。在分析26個玉米基因體的研究中 [13] 發現了更多的 SV 參與疾病的發生而不是賦予重要的農藝性狀。
總結:
三代定序有利於提高變異單倍型的連續性、準確性和範圍,評估複雜的小變異,並已應用於尋找與疾病相關的等位基因 [14] [15] [16],相信隨著該領域的不斷進步,可能會有更多的研究跟進。
參考資料:
回上一頁
? 為什麼西方人皮膚是白的?
? 為什麼人口平均身高最高的國家幾乎都在歐洲?
? 為什麼心臟病、糖尿病與氣喘等常見的慢性病,發生在非裔美國人的比率高於非拉丁裔的白人?
… (以下省略一百個問題,絕對是因為篇幅問題,不是小編偷懶喔)
最後要問:
為什麼每個族群看起來那麼不同卻都使用同一個參考基因體呢?
最後一個問題也是現在各國積極推動大型族群研究的原因
--- 建立該國的人群資料庫
背景及挑戰:
事實上,早在 1948 年開始,世界各地陸續開始成立國家型人體生物資料庫 (圖一),包含美國、英國、中國、日本、韓國等國家,期望這些資料庫能發掘族群特有的遺傳變異、對於複雜的遺傳疾病能更好理解以及快速發現,從而實現精準化治療。

(圖一) 圓形表示全球基因體資料庫; 方形表示相關研究 [1]
以前的族群研究,包括全基因體關聯研究 (GWAS),並未能詳盡地描述人類特徵和遺傳疾病的相關性 [2]。關於這種“遺傳性缺失”的來源有很多猜測,通常指向結構變異 (SV) 和罕見變異。
在人類基因體中,SVs 所影響的核苷酸總數比單核苷酸變異 (SNV) 數量多得多。但直到目前為止,此類族群研究仍主要依賴於二代定序技術完成,該技術產生的讀長範圍為 25 bp 至 600 bp。
然而,二代定序的短讀長在重複區域方面遭遇到嚴重限制。DNA 重複序列作為基因體單元促進了 SV 的形成,同時也由於序列無法準確對齊而阻礙了 SV 的發現。即使在非重複區域中,二代定序由於技術限制也容易遺漏插入等變異(尤其是長度超過定序讀長的等位基因) 或其他修飾(例如甲基化)。隨著三代定序的成熟,越來越多研究開始將三代定序作為基因計畫的技術平台。
使用三代定序之大型研究:
瓶中基因體 (GIAB) 和人類基因體結構變異 (HGSV) 聯盟結合了多種技術來全面表徵人類基因體的結構變異,不約而同表示,三代定序在結構變異識別的能力已遠優於二代序和其他方法(例如 arrays)[3] [4] 。這些研究強調,通過三代定序可以發現很大一部分隱藏的變異。
已經有越來越多的大型研究納入三代定序作為發掘 SVs 的工具,從 2019 年到 2021 年使用三代定序且超過 5 個樣本的研究統計如圖二; 詳細列表於表一。

(圖二) 圓點大小表示基因體大小; 顏色表示發掘遺傳變異的方法 [5]
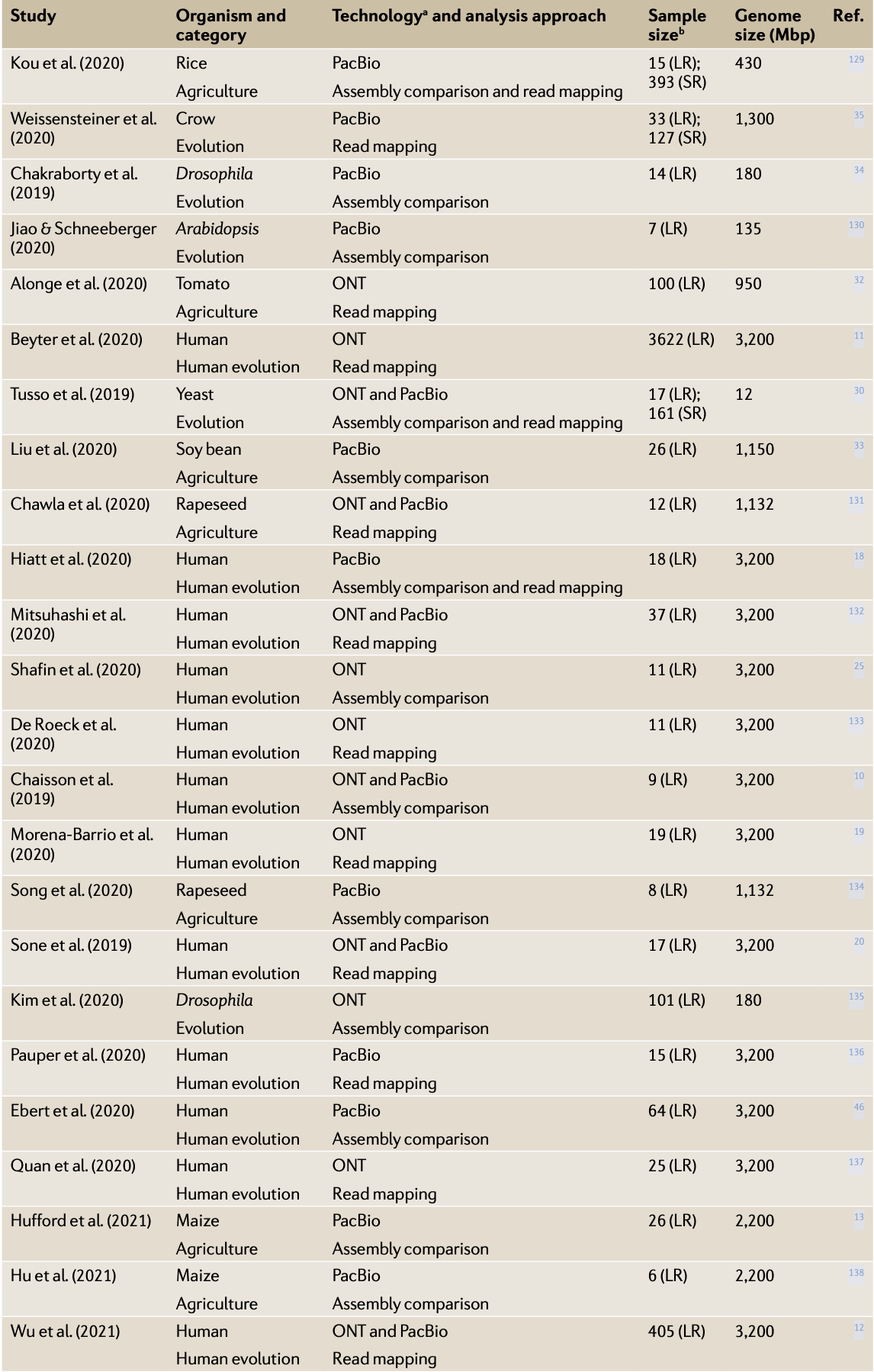
(表一) 使用三代定序之族群研究列表 [5]

(表一) 使用三代定序之族群研究列表 [5]
定序策略:
在大規模人群定序項目開始時,需要考慮多種具有特定預算要求的常用策略(圖三)。
下列討論三種主要策略,以達成不同的規模和預算,但須注意其對檢測遺傳變異的辨別率會產生影響。

(左) 全覆蓋策略(Full coverage):
所有樣本都以中 ( >12x 定序深度) 到高 (>40x 定序深度) 的三代定序深度進行定序。儘管其成本為這三種方法中最高,但其優勢在於全面性、高水平的分辨率、研究設計的簡單性和相對簡單的計算工作流程。
假設: 20x 的覆蓋度對 2,500 人進行定序,需要 150 Tb的數據量。
(中) 混合覆蓋策略 (Mixed coverage) :
代表樣本 (如: 亞種) 以中到高覆蓋度 (30x 定序深度) 進行三代定序,其餘樣本使用低覆蓋度 (>5x 定序深度) 進行三代定序(類似於最初的 1000 Genomes 項目)。混合覆蓋策略雖然比全覆蓋策略便宜,但仍然可以實現較高的整體檢測靈敏度,因此特別適用於人數眾多或預算有限的研究。但是,分析具有較高挑戰,特別是在跨多個樣本實現準確基因型預測、或是區分體細胞與雜合種系變異等。此外,混合覆蓋策略偏向發現常見等位基因,許多稀有等位基因可能會被遺漏。
假設: 30x 的覆蓋度對 200 人定序,其餘個體以 8x 覆蓋度進行定序,僅需要 73 Tb 的數據量,是全覆蓋策略的一半。
(右) 混和定序策略 (mixed sequencing approach)
一部分樣本 (10~20%) 以三代定序進行中到高覆蓋度定序,其餘樣本使用二代定序。採用哪種方法的決定將影響檢測人群中常見(紅色符號)或罕見(灰色符號)變異的能力。該決定還取決於可用預算、現有數據和樣本 DNA 可用性。混和定序策略的原理類似於混合覆蓋策略,可有效闡明易患癌症的種系 SV,其中二代定序用於驗證 SV。有些研究團隊使用 SVCollector [6] 自動選擇樣本進行三代定序,其識別的 SV 以現有的二代定序數據集中對 SV 的斷點坐標進行基因分型(例如,插入)。通過這種方式,可以獲得已識別變體的穩健等位基因頻率,但依然可能會遺漏其他樣本中包含的稀有變體。
假設: 30x 的覆蓋度對 200 人定序,其餘個體使用二代定序數據,僅需要 18 Tb 的數據量,是最經濟實惠的選項。
最新案例:

迄今為止,最大規模且以人類為中心的三代定序研究於 2021 發表於 Nature Genetics [7],本篇研究利用 Nanopore 平台檢測 3,622 位冰島人基因體多樣性,發現每位冰島人平均擁有 22,636 個結構變異,包含了 13,353 個插入及 9,474 個缺失變異。這些變異中,含有 133,886 個可信的等位基因結構變異。接著,在 166,281 位冰島人中,驗證這些變異與疾病之間的關係。研究結果發現,擁有 PCSK9 基因第一個外顯子缺失者,其低密度脂蛋白的濃度較一般人平均濃度低。而另一個基因 ACAN,有長度為 57 bp 的多個重複序列,包含了 11 個等位基因型,其重複次數與個體身高有著顯著相關。這篇研究證實了,三代定序長讀長的特點,可以有效的在族群間利用全基因體定序找到有意義的結構變異,甚至解密這些結構變異對於人類表型的影響。[8]
當然,除了本篇的冰島人基因體定序計畫,還有阿布達比的 100,000 人基因定序計畫、NIH All of Us 研究計劃和 NIH 阿茲海默症和相關癡呆症中心 (CARD) 在美國和中國、阿布扎比和卡塔爾的類似研究,都可以從中看出三代定序的應用層面逐漸擴大與普及。
除了人類層面的研究之外,三代定序已在族群規模上應用,以發現與作物 [9] [10]、果蠅 [11] 和鳴禽 [12] 的表型相關的結構變異。在分析26個玉米基因體的研究中 [13] 發現了更多的 SV 參與疾病的發生而不是賦予重要的農藝性狀。
總結:
三代定序有利於提高變異單倍型的連續性、準確性和範圍,評估複雜的小變異,並已應用於尋找與疾病相關的等位基因 [14] [15] [16],相信隨著該領域的不斷進步,可能會有更多的研究跟進。
參考資料:
- Carress, H., Lawson, D.J. & Elhaik, E. Population genetic considerations for using biobanks as international resources in the pandemic era and beyond. BMC Genomics 22, 351 (2021). https://doi.org/10.1186/s12864-021-07618-x
- Patron, J., Serra-Cayuela, A., Han, B., Li, C. & Wishart, D. S. Assessing the performance of genome-wide association studies for predicting disease risk. PLoS ONE 14, e0220215 (2019).
- Zook, J. M. et al. A robust benchmark for detection of germline large deletions and insertions. Nat. Biotechnol. 38, 1347–1355 (2020).
- Chaisson, M. J. P. et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 10, 1784 (2019).
- De Coster, W., Weissensteiner, M.H. & Sedlazeck, F.J. Towards population-scale long-read sequencing. Nat Rev Genet 22, 572–587 (2021). https://doi.org/10.1038/s41576-021-00367-3
- Ranallo-Benavidez, T. R. et al. Optimized sample selection for cost-efficient long-read population sequencing. Genome Res. https://doi.org/10.1101/ gr.264879.120 (2021). This article describes a method for optimized sample selection given an existing variation catalogue.
- Beyter, D., Ingimundardottir, H., Oddsson, A. et al. Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat Genet 53, 779–786 (2021). https://doi.org/10.1038/s41588-021-00865-4
- https://www.facebook.com/groups/883166995753861/posts/970577807012779
- Alonge, M. et al. Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 182, 145–161.e23 (2020). This study reports the population-scale sequencing for a plant (tomato) and details the impact of the detected variation on phenotypes.
- 33. Liu, Y. et al. Pan-genome of wild and cultivated soybeans. Cell 182, 162–176.e13 (2020).
- Chakraborty, M., Emerson, J. J., Macdonald, S. J. & Long, A. D. Structural variants exhibit widespread allelic heterogeneity and shape variation in complex traits. Nat. Commun. 10, 4872 (2019).
- genomics of structural variation in a songbird genus. Nat. Commun. 11, 3403 (2020). A large-scale sequencing study in crows highlights segregation of structural variation in natural populations.
- Hufford, M. B. et al. De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. bioRxiv, https://doi.org/10.1101/2021.01.14.426684 (2021).
- Hiatt, S. M. et al. Long-read genome sequencing for the diagnosis of neurodevelopmental disorders. bioRxiv https://doi.org/10.1101/2020.07.02.185447 (2020).
- de la Morena-Barrio, B. et al. Long-read sequencing resolves structural variants in SERPINC1 causing antithrombin deficiency and identifies a complex rearrangement and a retrotransposon insertion not characterized by routine diagnostic methods. bioRxiv https://doi.org/10.1101/2020.08.28.271932 (2020).
- Sone, J. et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 51, 1215–1221 (2019).
圖爾思生物科技 / 微生物體研究中心
吳雁韻 文案



