19
2021.11
【蛋白質體學】由Quantification Proteomic邁向精準醫療時代 !
人們對治療的概念,自人類基因解碼的大數據時代開始走向「個人化(personalized)」的精準醫學(Precision Medicine)。打破以往相同疾病之病患僅採取相同治療手段、用藥,精準醫學將個人資料如性別、身高、體重、種族、基因檢測、蛋白質檢測、代謝檢測、過去病史、家族病史等,經由人體的生物資訊資料庫進行比對及分析,從中找出最適合病患的治療方法與藥品,以達到藥物最大療效與最小的副作用。
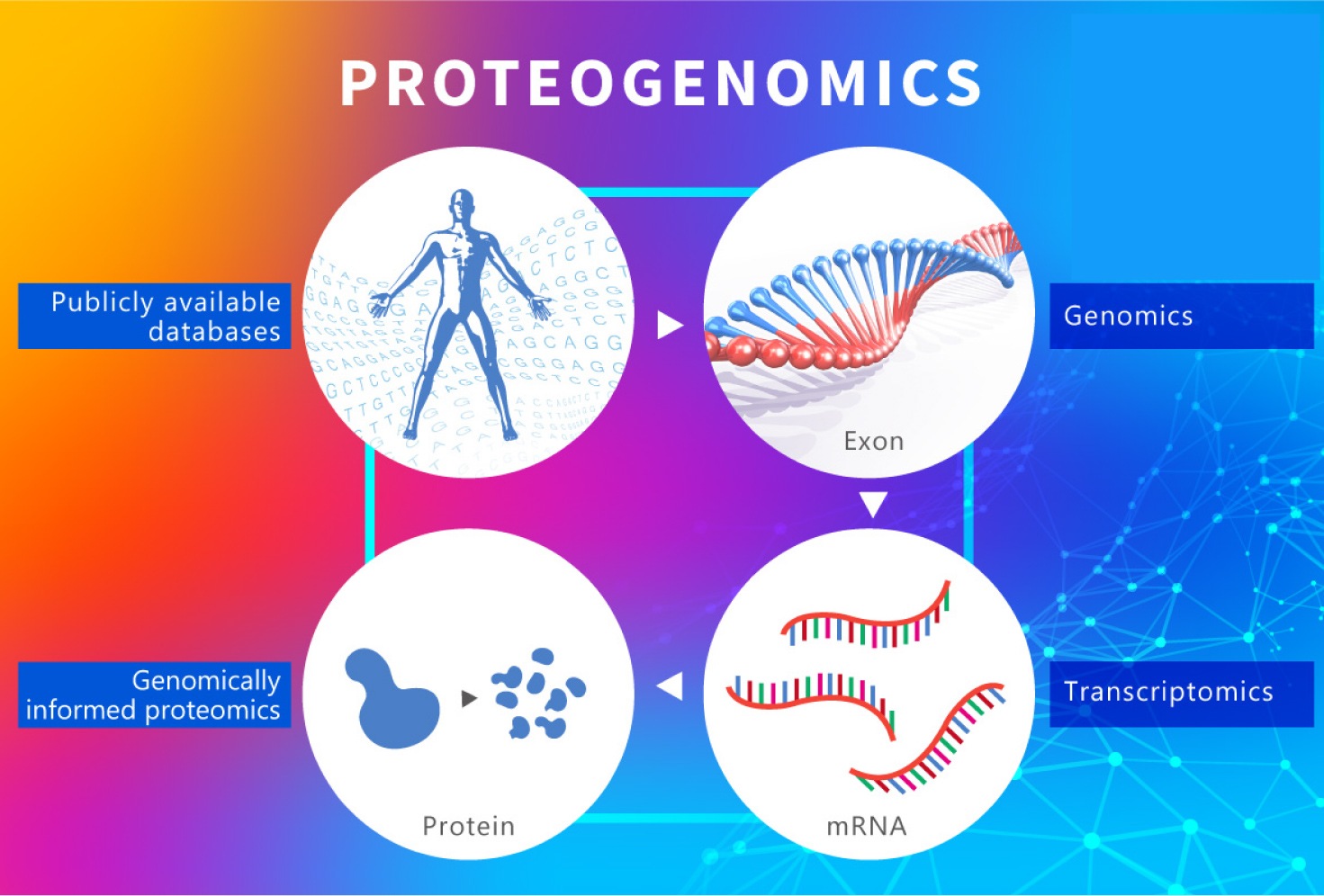
蛋白基因組學 (Proteogenomics)是近年精準醫療的熱門平台 ¹。Proteogenomics是將基因組、轉錄組、蛋白組,以及轉譯後修飾組的大數據整合,從多分子層面的大數據重新定義疾病的分型、發掘潛在的治療靶點,最終在蛋白層次上發現與驗證腫瘤相關基因突變、表達變化及關鍵分子調控機制,以進行精準化用藥指導和藥物開發。(圖一):
蛋白基因組學 (Proteogenomics)是近年精準醫療的熱門平台 ¹。Proteogenomics是將基因組、轉錄組、蛋白組,以及轉譯後修飾組的大數據整合,從多分子層面的大數據重新定義疾病的分型、發掘潛在的治療靶點,最終在蛋白層次上發現與驗證腫瘤相關基因突變、表達變化及關鍵分子調控機制,以進行精準化用藥指導和藥物開發。(圖一):
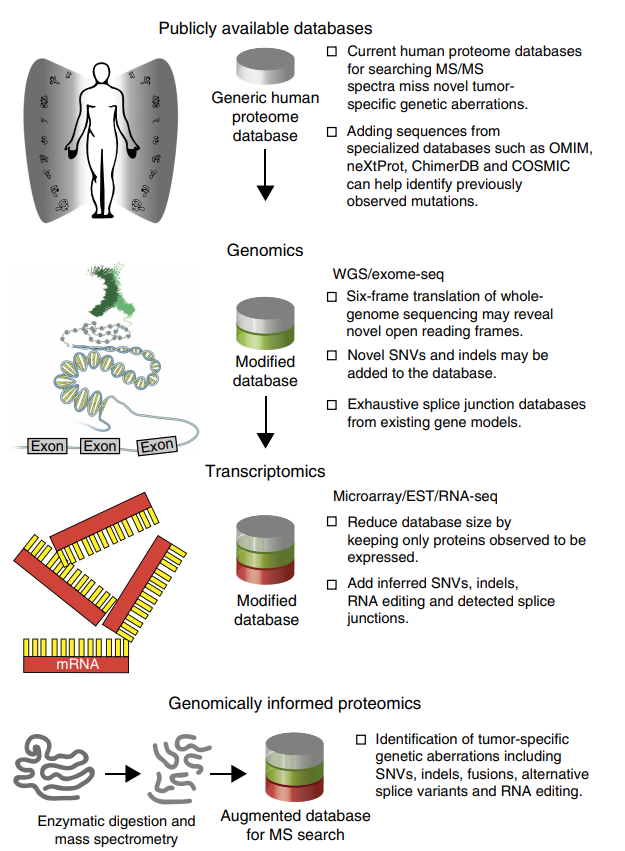
(圖一)
Alfaro, J., Sinha, A., Kislinger, T. et al. Onco-proteogenomics: cancer proteomics joins forces with genomics. Nat Methods 11, 1107–1113 (2014).
https://doi.org/10.1038/nmeth.3138
2016年美國癌症登月計劃(Cancer Moonshot 2020)讓精準醫學時代正式開啟;同年,美國VA、DoD、NCI三部門聯合宣佈將建立第一個同時進行基因資訊和蛋白資訊表徵的醫學系統,把基因組和蛋白質組作為常規檢測手段,對癌症病人進行個人化蛋白基因組(Proteogenomics)表徵,為更精準的用藥提供指導。
發掘治療靶點的過程大致分成兩個部分:1) 高通量鑑定、2) 定量差異;其中在蛋白質體組學研究中,圖爾思SuperLab Proteomic Service所提供的標記定量技術iTRAQ(Isobaric Tags for Relative and Absolute Quantitation)和TMT(Tandem Mass Tags)無疑占據了半壁江山,是定量蛋白質組學中最經典、應用性佳的高通量篩選技術。
iTRAQ與TMT兩者的原理基本相同,分別由AB Sciex、ThermoFisher Scientific所開發,且標籤的標記數量不同。兩者皆包括三部分 (圖二):
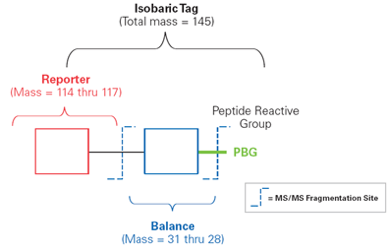
(圖二)
- 報告基團(reporter group)質量分別為分子量差1的基團。
- 平衡基團(balance group)質量分別為分子量差1的基團,使得iTRAQ試劑報告基團和平衡分子的總分子量均為145Da,無論使用哪種iTRAQ試劑,不同同位素標記同一肽段後在一級質譜中,分子量完全相同,呈現的都是同一波峰值。
- 肽反應基團(peptide reactive group)是將reporter group與肽N端及Lysine側鏈連接,從而將報告基團和平衡基團標記到peptide上,幾乎可以標記樣本中所有蛋白質。
目前iTRAQ有4-plex或8-plex同位素標籤,最多可以一次標記8個樣本,進行同步定量比較;而TMT則具有16 plex同位素標籤,最多可以一次標記16個樣本。
當面臨大規模的臨床研究,樣本的數量是成百上千的時候,該如何運用標記定量技術呢?以下為當前研究² ³所採用的實驗設計方法:
1. 設定定量分析的批次 (plex set)
(plex)內的樣本除了Control,盡量每一個樣本都具有不同的實驗意義,例如:疾病、用藥等。
2. 建立pooled global internal standards (GIS)
避免分析批次之間因Control樣本不同而造成差異,建議將所有的Control樣本合併成一個樣本,即GIS,而每次批次上機與分析時皆採同一GIS;在進行合併之前,也建議先分析每個Control樣本的蛋白質總量與定量,以確保Control之間的數據一致性。
3. 樣本的實驗分組意義與生物性重複
呈1.,先針對樣本的實驗意義進行分組,若不同樣本具有生物性重複,則可作為批次設定的依據,舉例說明:50個樣本,實驗分組為Normal、Disease、Disease + Treatments (8種),五重複,則建議採用10 plex,五個分析批次,每批次都應包含不同實驗設計組別;最後五個批次個別分析出的差異蛋白,取交集或聯集,篩選出具重複意義的差異蛋白。
4. 分析批次的數據一致性
整合批次間的數據,則必須考慮到實驗再現性;可利用每一定量分析批次 (TMT batch) 的the number of identified proteins、the number of identified proteins來界定批次間數據的一致性。建立合理的取樣模式與實驗分組是臨床或大規模研究的第一步,希望以上對所有正在、預計進行大規模目標蛋白探究的您有幫助。除本次分享,我們也會持續地閱讀文獻、吸收新知,與台灣的研究工作者們分享;如喜歡本次的文章或對定量蛋白質體學服務有興趣,請點擊以下連結,取得更多訊息;或與我們的團隊聊聊,討論更多研究主題的可能性:
Reference
1. Alfaro, J. A., Sinha, A., Kislinger, T. &Boutros, P. C. Onco-proteogenomics: Cancer proteomics joins forces with genomics. Nature Methods vol. 11 (2014).
2. Oh, S. et al. Integrated pharmaco-proteogenomics defines two subgroups in isocitrate dehydrogenase wild-type glioblastoma with prognostic and therapeutic opportunities. Nat. Commun. 11, (2020).
3. Chen, Y. J. et al. Proteogenomics of Non-smoking Lung Cancer in East Asia Delineates Molecular Signatures of Pathogenesis and Progression. Cell 182, (2020).



