23
2017.03
10x Genomics 最新研究成果─鑑定疾病基因體複雜的結構變異
10x Genomics 定序技術平台發表至今,以迅雷不及掩耳之勢強勢進軍定序領域,基於獨特的GemCode平台及linked-read定序使其可獲得極長片段遺傳資訊,從而進行染色體結構變異及單倍型 (Haplotype)檢測。
自2016年發表的對生殖系統和癌症基因體進行單倍型分析和結構變異檢測[1],探索罕見基因剔除對人類健康和族群的影響[2],再到結合BioNano Genomics組裝人類基因體資訊[3 ]和結合PacBio等技術組裝最連續的韓國人基因體[4]等多項研究成果,均表明了linked-read定序可以有效應用於結構變異檢測、Haplotype檢測及基因體組裝。近期,一篇利用10X Genomics技術進行疾病基因體結構變異檢測的文章揭示了疾病基因體結構變異的複雜性與多樣性,接下來小編將對此篇文章進行解析。
Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome[5]
期刊:Genome Biology 發表日期:2017.3.6 IF:11.3
liWGS中發現高度異質性11,735個不同的SVs,共檢測到 436,741 個SV觀測值,平均每個基因體有637個大型SVs。結合5種正交法 (Orthogonal approach)驗證SV發現具有10.6%偽陽性(FDR) 和5.9%偽陰性(FNR),其中驗證率最高的為佔多數的複雜結構變異 (complex SV, cxSV; 2.6% FDR )和典型deletions (5.3% FDR)。最低的為insertions (22.9% FDR),因其大部分插入片段比liWGS中的解析度小,除去這一類型,總體FDR為9.1%。重要的是,發現16.8%的 SVs 為均衡或複雜的SV,強調了當單獨分析典型CNVs時會忽視每個基因體中可觀的大型SV數目。分析還發現,10.9%的病患至少含有1個非常大型的罕見 SV (≥1 Mb;variant frequency (VF) < 1%),暗示罕見SV在個體基因體的頻率差異。
2. 新生SV位點和染色體重組複雜性
本研究檢測到的SV其中38.1%(4233 / 11,108)未曾在資料庫中出現,特別是在cxSVs檢測中幾乎為新發現的SV,其中50.2% 的cxSVs至少一個斷點在先前研究中被發現,但可能錯誤的歸類到典型SV中。值得注意的是,97.4% 的 cxSVs得到驗證。但是,由於liWGS解析度的限制,研究者預測可能會低估這些變異及其整體結構的複雜性,這是因為liWGS 無法獲知SV斷點的微觀複雜度以及對cxSVs內小的變異(< 5 kb) 的檢測能力有限。總之,這些數據顯示,人類中的大型cxSVs比先前認識到的更豐富且更加多樣化。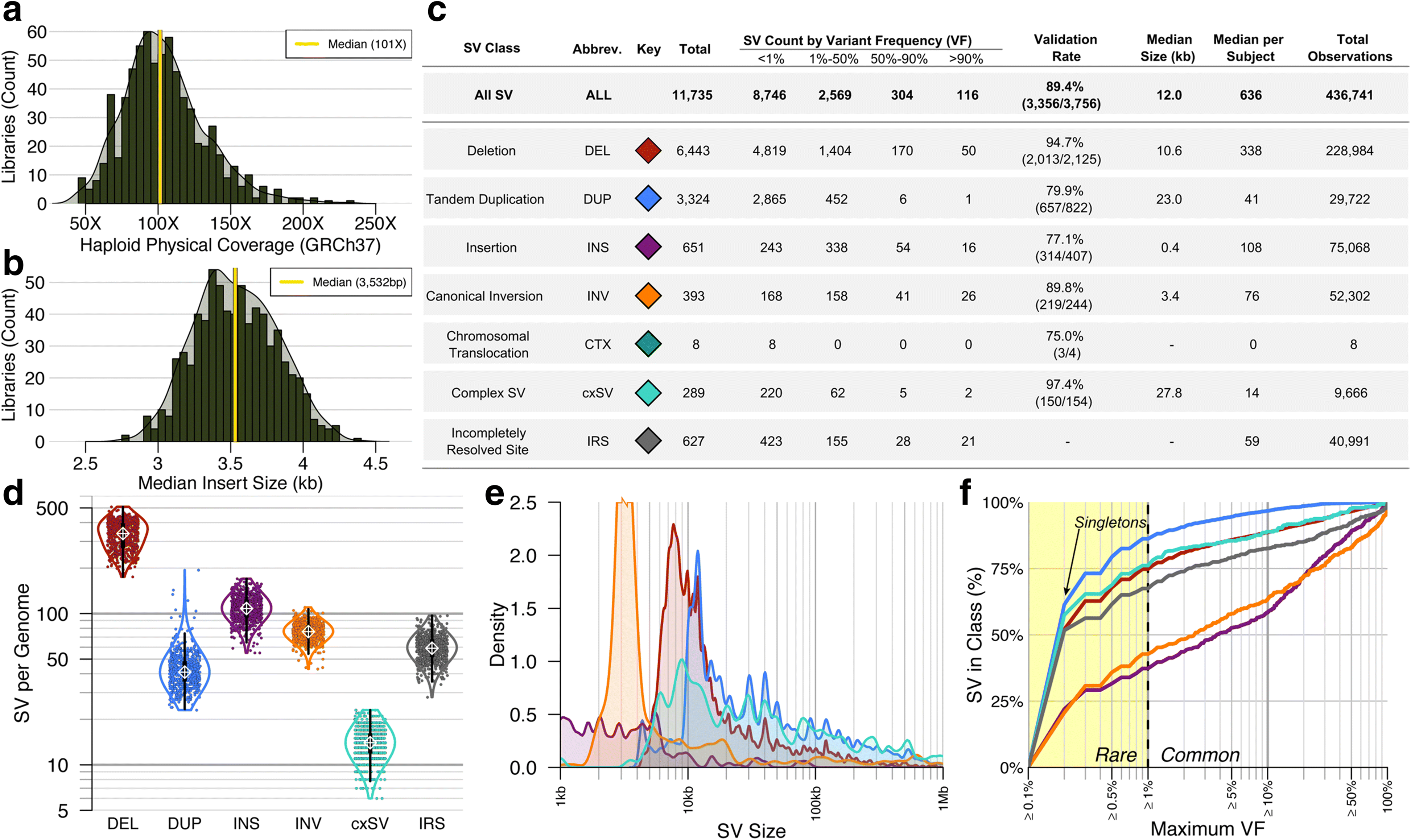
3. 定義及對比復發性cxSV的16個不同的亞類 (subclass)
本研究中新的大型cxSVs頻率可以進一步表示病患突變圖譜的特性。發現42.6% 的cxSVs 具多態性 (polymorphic ),平均每個病患有14個大型cxSVs,表明了cxSV是大部分人類基因體存在的變異類型。對復發性和相對常見的cxSV分為16個獨特的亞類,每個cxSV亞類至少在5個病患中存在並具有獨特的變異等位基因結構。這些亞類大部分為不均衡染色體倒置 (Chromosome inversions),因此大部分cxSV至少含有一個反向片段。 CNV-flanked inversions 中包含了最大的一組cxSVs (77.2%),其中複雜的重複相比缺失更大型且更罕見。相對於涉及2個及以上的染色體重組,大部分為染色體內cxSVs。綜合以上,在liWGS 解析度下,16種cxSV亞類展現了對人類cxSV數據的補充。
4. 典型及複雜的染色體倒置變異豐度
在liWGS中檢測到平均每個人有87個染色體倒置變異,發現其中12.6% 是複雜的染色體倒置變異,這些複雜的染色體倒置變異比常見的更大型,並且在罕見變異中顯著富集( VF < 1%),暗示了相對來說染色體倒置變異受到更大的選擇壓力,這種趨勢可能是與SV頻率及片段大小有一定的聯繫。大型的染色體倒置不易在生殖細胞系中存在,可能是由於其可增加有害性後果或阻礙重組發生。本研究在每個基因體中檢測到的染色體倒置變異數目比千人基因體樣本中多2倍。總體來說,此研究中觀察到的典型和複雜的染色體倒置變異,暗示了此變異亞類可通過提供長片段基因體結構資訊的定序技術獲得。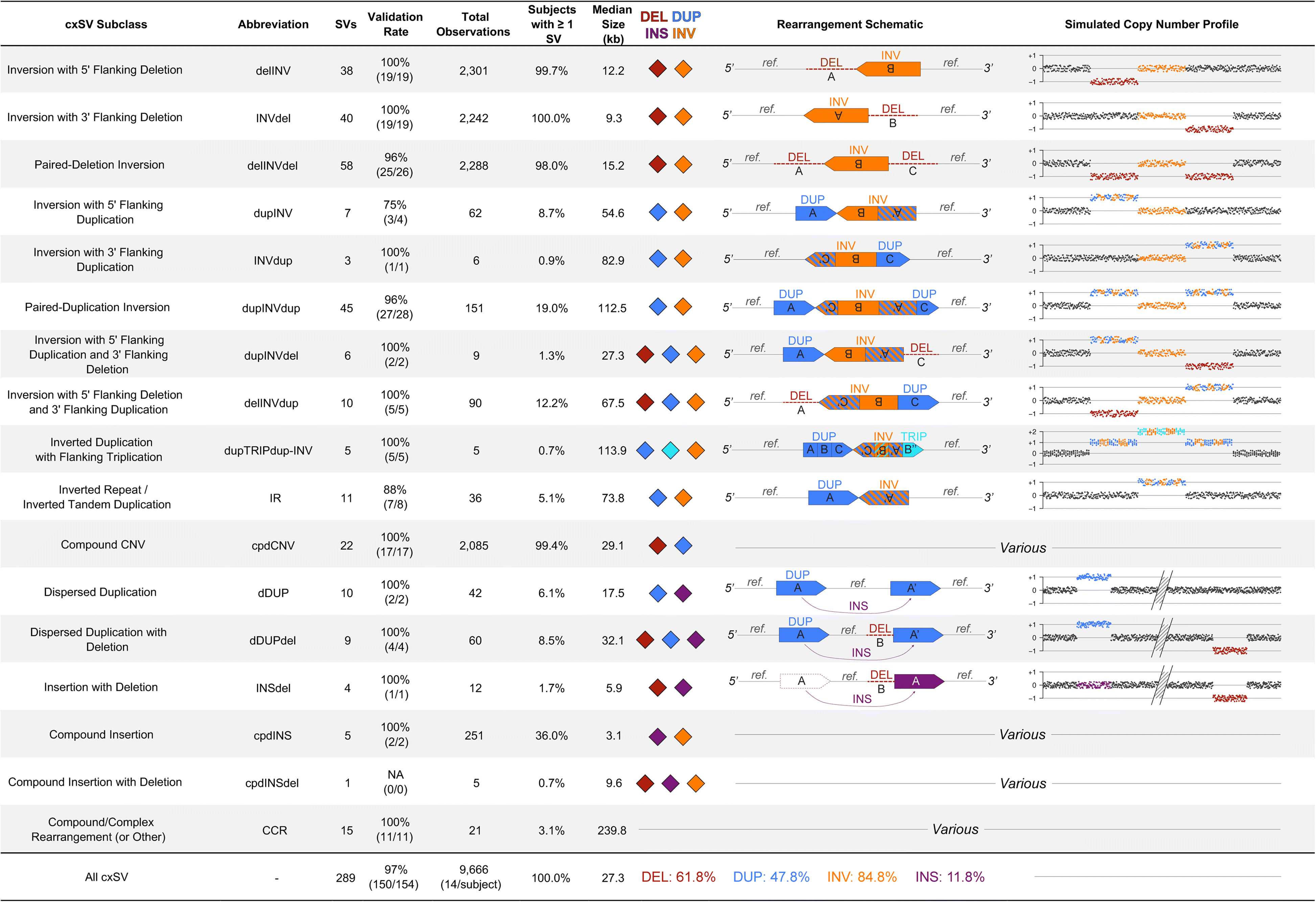
5. 使用linked-read WGS鑑定cxSV
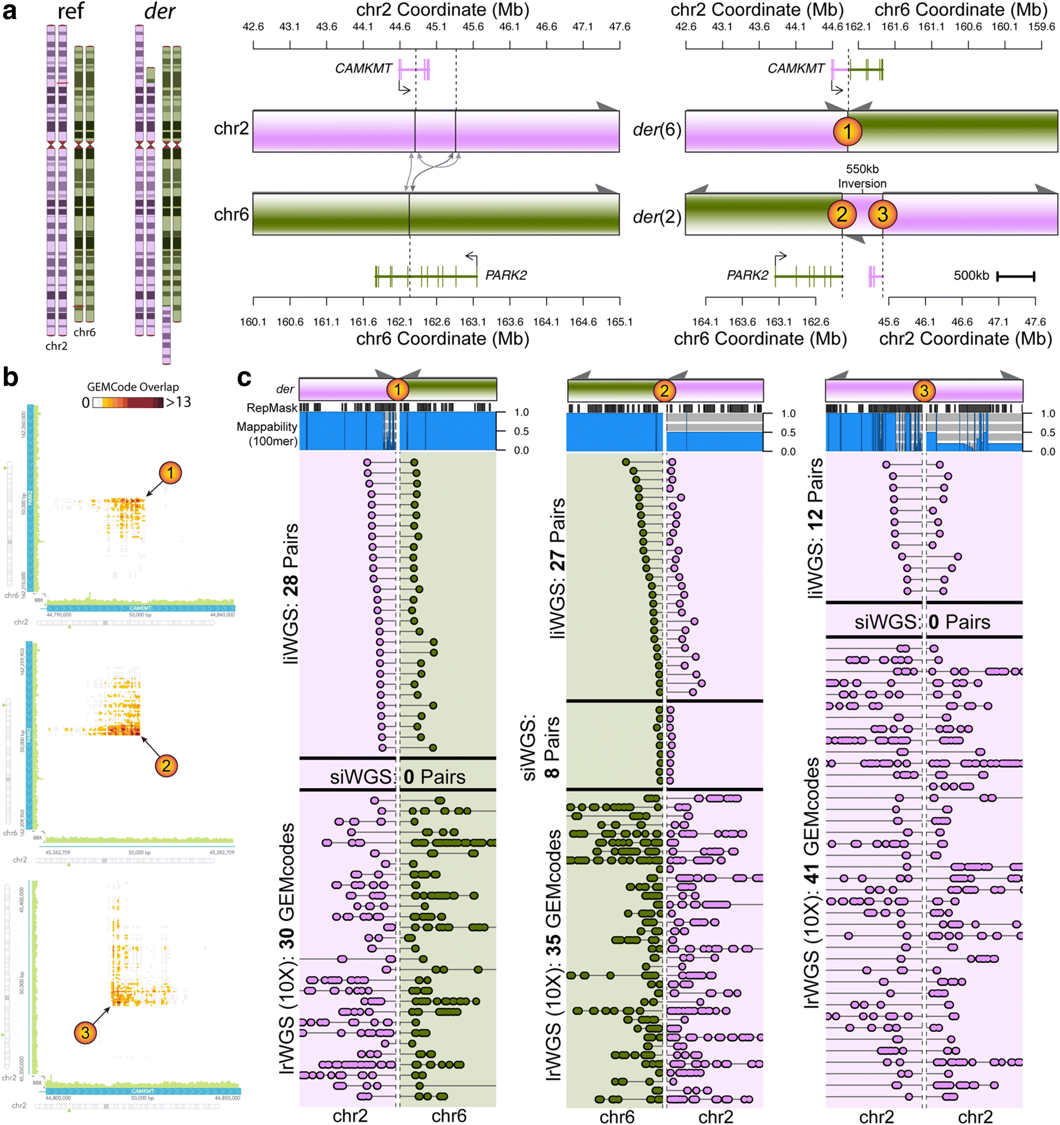
為檢驗在3例病患中利用liWGS檢測出但未通過正交法驗證的大型的罕見cxSVs,使用10X Genomics公司linked-read WGS (lrWGS)對此3例病例和其雙親父母進行定序,平均定序深度為31.1X。最終驗證了每個大型cxSV的所有斷點。值得注意的是其中包括了1名ASD 患者的1個全新未發現複雜染色體易位,此染色體易位在liWGS預測中涉及550 kb 的倒位序列和3個斷點,其中2個斷點在傳統方法( PCR和Sanger)和siWGS中未通過驗證。所有的三個斷點通過10X Genomics 的104個獨立的 lrWGS分子得到驗證與定相 (phasing),揭示了 PARK2 和 CAMKMT 基因受到破壞。這些數據進一步表明,對於解決大型的複雜染色體異常,提供長片段結構資訊的技術十分有價值,且在更大的樣本量中需要綜合分析方法以提高lrWGS相比siWGS、liWGS或其他新興技術的SV檢出量。
6. 罕見SVs 顯示多種有害的生物學特徵
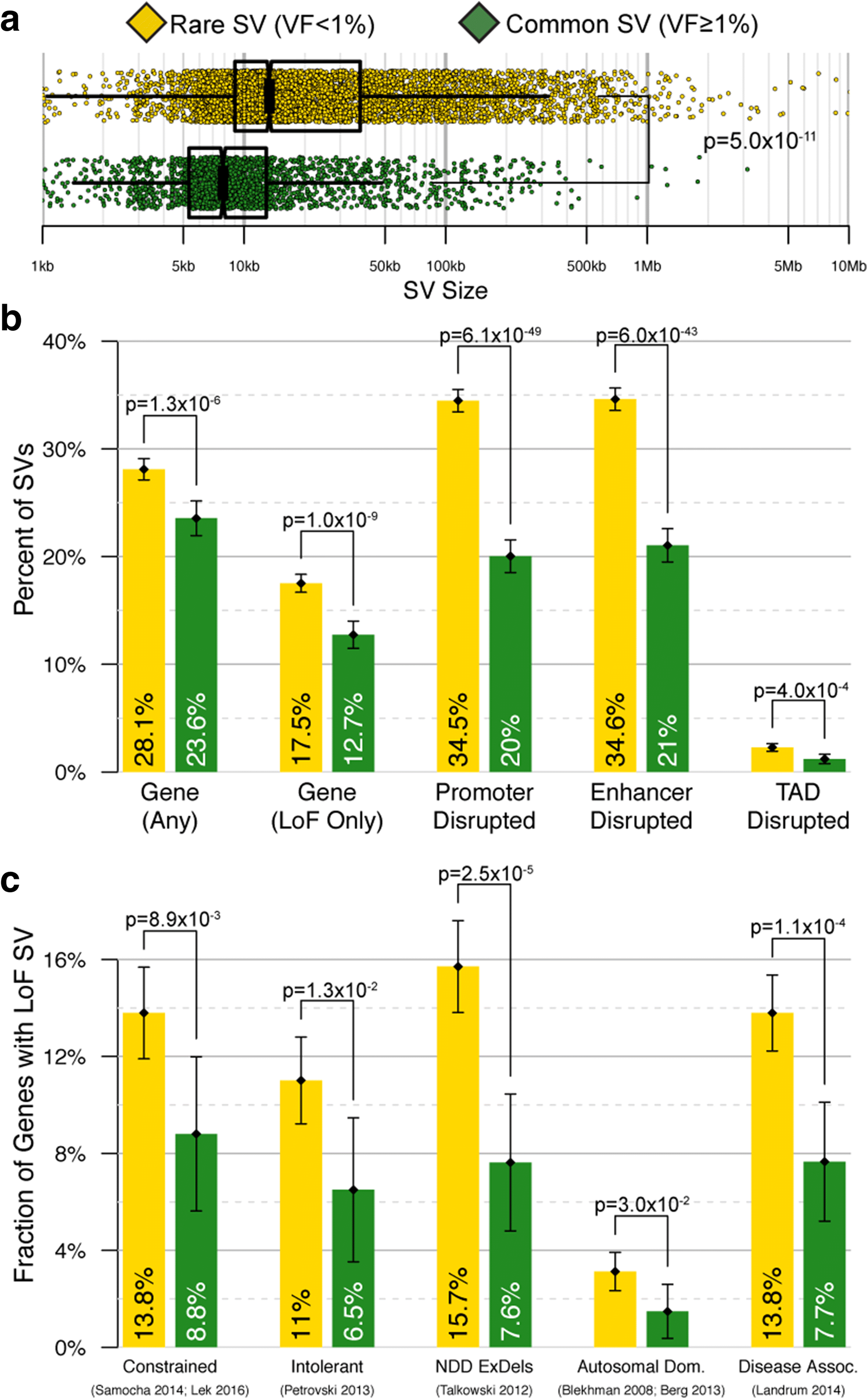
與罕見編碼位點突變趨勢一致,相對於常見多態性SVs,罕見SVs似乎更有危害性,此研究中罕見SVs相對常見SVs更大型,這與千人基因體計劃觀察到的結果一致。在此研究中,由罕見低頻率SV截斷的基因集大約兩倍富集在疾病相關基因、功能突變不耐受基因以及外顯子缺失基因,顯示了對有害SV的選擇壓力。最後,鑑定了十個特異性基因座,其顯著富集於罕見SV並超出全基因體預期豐度,其中5個基因涉及神經發展疾患 ( PARK2、IMMP2L 、CTNNA3、CYFIP1、PTPRT )。
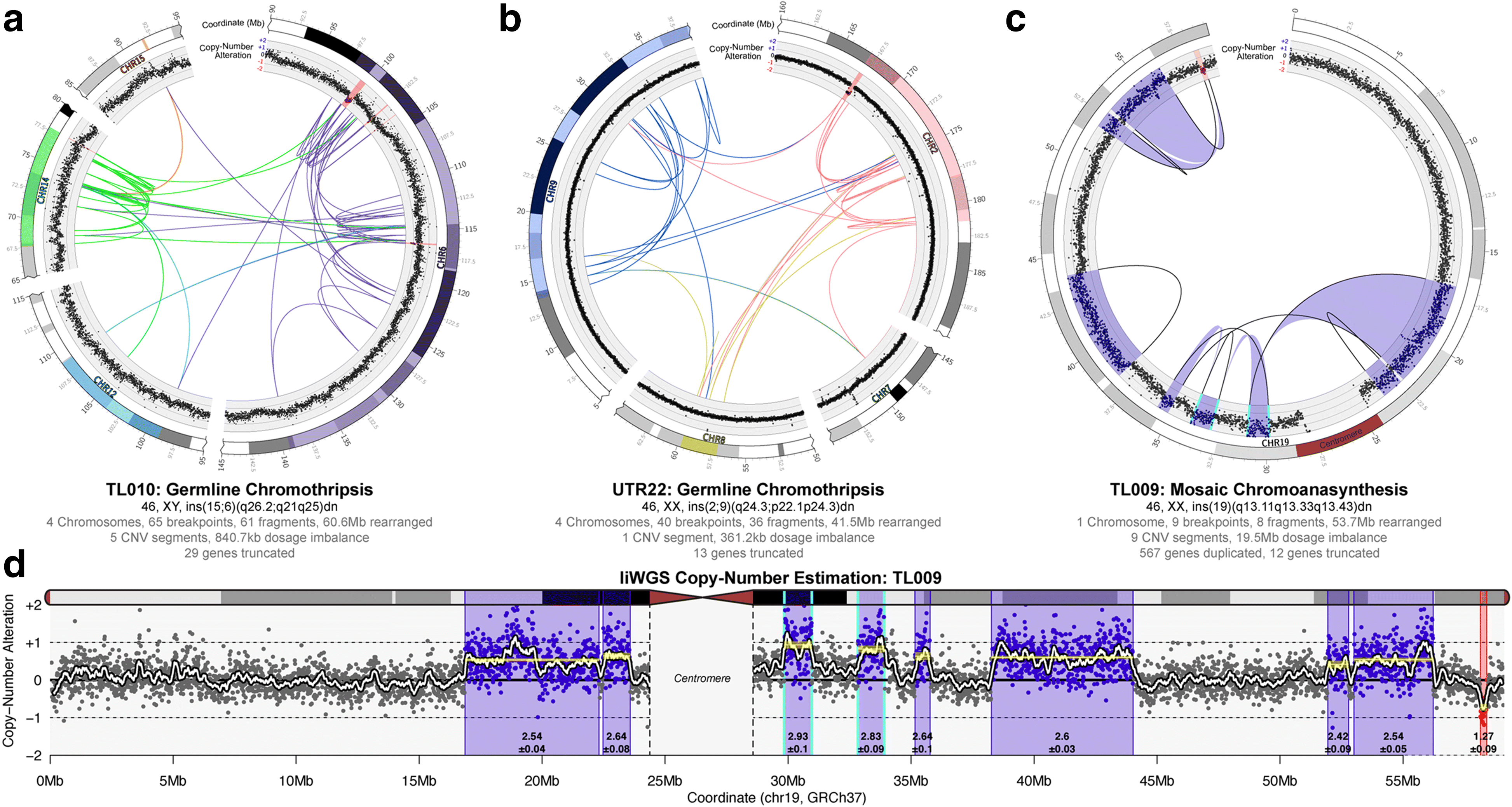
7. 人類發育異常中的極端chromoanagenesis
最具危害性的cxSV亞類稱為chromoanagenesis ,根據對chromoanagenesis了解及本研究的發現,查詢文章報導發現其在受影響的個體中幾乎都為新生chromoanagenesis。基於這些認知,研究者對額外的3例發育異常的病患進行liWGS,通過臨床核型分析 (Karyotypeing)發現了大型的新生易位插入片段,研究者懷疑存在更複雜的染色體重組。 3例病患中的chromoanagenesis檢測情況表明極端chromoanagenesis可能在生殖細胞系中出現,但常導致接近中性劑量衍生物,不均衡的chromoanasynthesis可能在體細胞出現,很可能存在暫時被打斷的一系列染色體重組中,且相對單個突變過程,其更類似chromoplexy複合突變過程。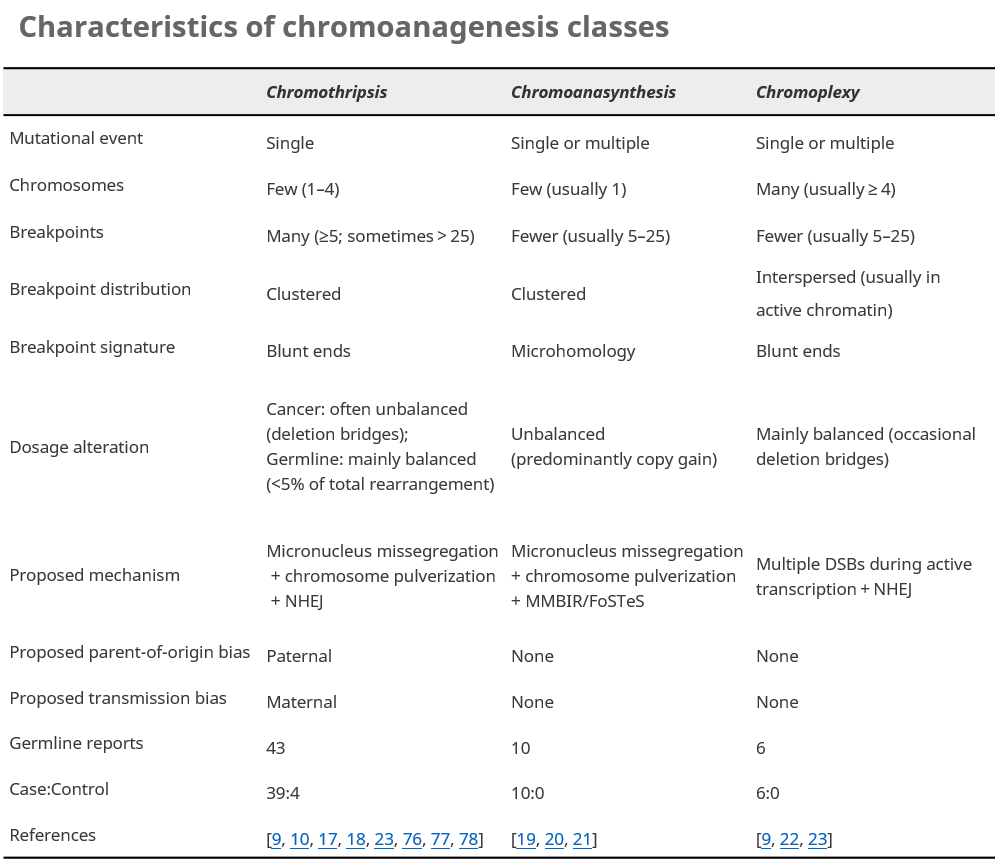
參考文獻
[1] ZhengG X, Lau B T, Schnalllevin M, et al. Haplotyping germline and cancer genomes using high-throughput linked-read sequencing[J]. Nature Biotechnology, 2016,34(3):303
[2] Narasimhan V M, Hunt K A, Mason D, et al. Health and population effects of rare gene knockouts in adult humans with related parents[J]. Science, 2016, 352(6284):474
[3] Mostovoy Y, Levysakin M, Lam J, et al. A hybrid approach for de novo human genome sequence assembly and phasing[J]. Nature Methods, 2016, 13(7):587
[4] Seo J S, Rhie A, Kim J, et al. De novo assembly and phasing of a Korean human genome[J]. Nature, 2016, 538(7624):243–247
[5] Ryan L. Collins, Harrison Brand, Claire E. Redin, et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome[J]. Genome Biology, 2017, 18(1) :36
回上一頁
自2016年發表的對生殖系統和癌症基因體進行單倍型分析和結構變異檢測[1],探索罕見基因剔除對人類健康和族群的影響[2],再到結合BioNano Genomics組裝人類基因體資訊[3 ]和結合PacBio等技術組裝最連續的韓國人基因體[4]等多項研究成果,均表明了linked-read定序可以有效應用於結構變異檢測、Haplotype檢測及基因體組裝。近期,一篇利用10X Genomics技術進行疾病基因體結構變異檢測的文章揭示了疾病基因體結構變異的複雜性與多樣性,接下來小編將對此篇文章進行解析。
Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome[5]
期刊:Genome Biology 發表日期:2017.3.6 IF:11.3
研究背景
結構變異 (Structural variation,SV)不僅影響基因體的結構且影響人類疾病的發生,但在疾病相關研究中仍未獲得較為完整的SV突變圖譜。本文對689例發育障礙病患進行疾病相關SV檢測,以期構築全基因體SV起始圖譜,作為未來研究疾病SV的參考基準。
研究方法

研究結果
1. 疾病基因體中SV的發現與檢驗liWGS中發現高度異質性11,735個不同的SVs,共檢測到 436,741 個SV觀測值,平均每個基因體有637個大型SVs。結合5種正交法 (Orthogonal approach)驗證SV發現具有10.6%偽陽性(FDR) 和5.9%偽陰性(FNR),其中驗證率最高的為佔多數的複雜結構變異 (complex SV, cxSV; 2.6% FDR )和典型deletions (5.3% FDR)。最低的為insertions (22.9% FDR),因其大部分插入片段比liWGS中的解析度小,除去這一類型,總體FDR為9.1%。重要的是,發現16.8%的 SVs 為均衡或複雜的SV,強調了當單獨分析典型CNVs時會忽視每個基因體中可觀的大型SV數目。分析還發現,10.9%的病患至少含有1個非常大型的罕見 SV (≥1 Mb;variant frequency (VF) < 1%),暗示罕見SV在個體基因體的頻率差異。
2. 新生SV位點和染色體重組複雜性
本研究檢測到的SV其中38.1%(4233 / 11,108)未曾在資料庫中出現,特別是在cxSVs檢測中幾乎為新發現的SV,其中50.2% 的cxSVs至少一個斷點在先前研究中被發現,但可能錯誤的歸類到典型SV中。值得注意的是,97.4% 的 cxSVs得到驗證。但是,由於liWGS解析度的限制,研究者預測可能會低估這些變異及其整體結構的複雜性,這是因為liWGS 無法獲知SV斷點的微觀複雜度以及對cxSVs內小的變異(< 5 kb) 的檢測能力有限。總之,這些數據顯示,人類中的大型cxSVs比先前認識到的更豐富且更加多樣化。

本研究中新的大型cxSVs頻率可以進一步表示病患突變圖譜的特性。發現42.6% 的cxSVs 具多態性 (polymorphic ),平均每個病患有14個大型cxSVs,表明了cxSV是大部分人類基因體存在的變異類型。對復發性和相對常見的cxSV分為16個獨特的亞類,每個cxSV亞類至少在5個病患中存在並具有獨特的變異等位基因結構。這些亞類大部分為不均衡染色體倒置 (Chromosome inversions),因此大部分cxSV至少含有一個反向片段。 CNV-flanked inversions 中包含了最大的一組cxSVs (77.2%),其中複雜的重複相比缺失更大型且更罕見。相對於涉及2個及以上的染色體重組,大部分為染色體內cxSVs。綜合以上,在liWGS 解析度下,16種cxSV亞類展現了對人類cxSV數據的補充。
4. 典型及複雜的染色體倒置變異豐度
在liWGS中檢測到平均每個人有87個染色體倒置變異,發現其中12.6% 是複雜的染色體倒置變異,這些複雜的染色體倒置變異比常見的更大型,並且在罕見變異中顯著富集( VF < 1%),暗示了相對來說染色體倒置變異受到更大的選擇壓力,這種趨勢可能是與SV頻率及片段大小有一定的聯繫。大型的染色體倒置不易在生殖細胞系中存在,可能是由於其可增加有害性後果或阻礙重組發生。本研究在每個基因體中檢測到的染色體倒置變異數目比千人基因體樣本中多2倍。總體來說,此研究中觀察到的典型和複雜的染色體倒置變異,暗示了此變異亞類可通過提供長片段基因體結構資訊的定序技術獲得。

為檢驗在3例病患中利用liWGS檢測出但未通過正交法驗證的大型的罕見cxSVs,使用10X Genomics公司linked-read WGS (lrWGS)對此3例病例和其雙親父母進行定序,平均定序深度為31.1X。最終驗證了每個大型cxSV的所有斷點。值得注意的是其中包括了1名ASD 患者的1個全新未發現複雜染色體易位,此染色體易位在liWGS預測中涉及550 kb 的倒位序列和3個斷點,其中2個斷點在傳統方法( PCR和Sanger)和siWGS中未通過驗證。所有的三個斷點通過10X Genomics 的104個獨立的 lrWGS分子得到驗證與定相 (phasing),揭示了 PARK2 和 CAMKMT 基因受到破壞。這些數據進一步表明,對於解決大型的複雜染色體異常,提供長片段結構資訊的技術十分有價值,且在更大的樣本量中需要綜合分析方法以提高lrWGS相比siWGS、liWGS或其他新興技術的SV檢出量。

與罕見編碼位點突變趨勢一致,相對於常見多態性SVs,罕見SVs似乎更有危害性,此研究中罕見SVs相對常見SVs更大型,這與千人基因體計劃觀察到的結果一致。在此研究中,由罕見低頻率SV截斷的基因集大約兩倍富集在疾病相關基因、功能突變不耐受基因以及外顯子缺失基因,顯示了對有害SV的選擇壓力。最後,鑑定了十個特異性基因座,其顯著富集於罕見SV並超出全基因體預期豐度,其中5個基因涉及神經發展疾患 ( PARK2、IMMP2L 、CTNNA3、CYFIP1、PTPRT )。

7. 人類發育異常中的極端chromoanagenesis
最具危害性的cxSV亞類稱為chromoanagenesis ,根據對chromoanagenesis了解及本研究的發現,查詢文章報導發現其在受影響的個體中幾乎都為新生chromoanagenesis。基於這些認知,研究者對額外的3例發育異常的病患進行liWGS,通過臨床核型分析 (Karyotypeing)發現了大型的新生易位插入片段,研究者懷疑存在更複雜的染色體重組。 3例病患中的chromoanagenesis檢測情況表明極端chromoanagenesis可能在生殖細胞系中出現,但常導致接近中性劑量衍生物,不均衡的chromoanasynthesis可能在體細胞出現,很可能存在暫時被打斷的一系列染色體重組中,且相對單個突變過程,其更類似chromoplexy複合突變過程。


研究結論
這項研究在人類疾病基因體中對SVs亞類的數量和多樣性提供了新的見解,並指出,染色體倒置變異比先前認識到的更加複雜。此研究檢測的變異模式擴展了先前在一般人群中SVs的圖譜,且SV的功能註釋闡述罕見的SV比普通的SV更可能擾亂編碼和調控非編碼區域。這些分析進一步表明,由罕見SV截短的基因更可能與疾病表現型相關。 3例病患呈現的chromoanagenesis情況表明極度複雜的均衡染色體重組在人類生殖細胞系中是可耐受的,且一些毀滅性的染色體重組可通過多個突變事件產生。這項研究強調需要詳細的SV的特徵以幫助解釋疾病人類基因體,而此研究數據可提供一個染色體倒置和cxSVs的參考圖譜。參考文獻
[1] ZhengG X, Lau B T, Schnalllevin M, et al. Haplotyping germline and cancer genomes using high-throughput linked-read sequencing[J]. Nature Biotechnology, 2016,34(3):303
[2] Narasimhan V M, Hunt K A, Mason D, et al. Health and population effects of rare gene knockouts in adult humans with related parents[J]. Science, 2016, 352(6284):474
[3] Mostovoy Y, Levysakin M, Lam J, et al. A hybrid approach for de novo human genome sequence assembly and phasing[J]. Nature Methods, 2016, 13(7):587
[4] Seo J S, Rhie A, Kim J, et al. De novo assembly and phasing of a Korean human genome[J]. Nature, 2016, 538(7624):243–247
[5] Ryan L. Collins, Harrison Brand, Claire E. Redin, et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome[J]. Genome Biology, 2017, 18(1) :36
圖爾思生物科技/ 諾禾致源文案
http://www.toolsbiotech.com/
(配圖來源於網路,侵刪)
http://www.toolsbiotech.com/
(配圖來源於網路,侵刪)



