12
2022.01
你。不只是你。總體基因體學揭開人體內宇宙 (下)
原創文章 引用請註明出處
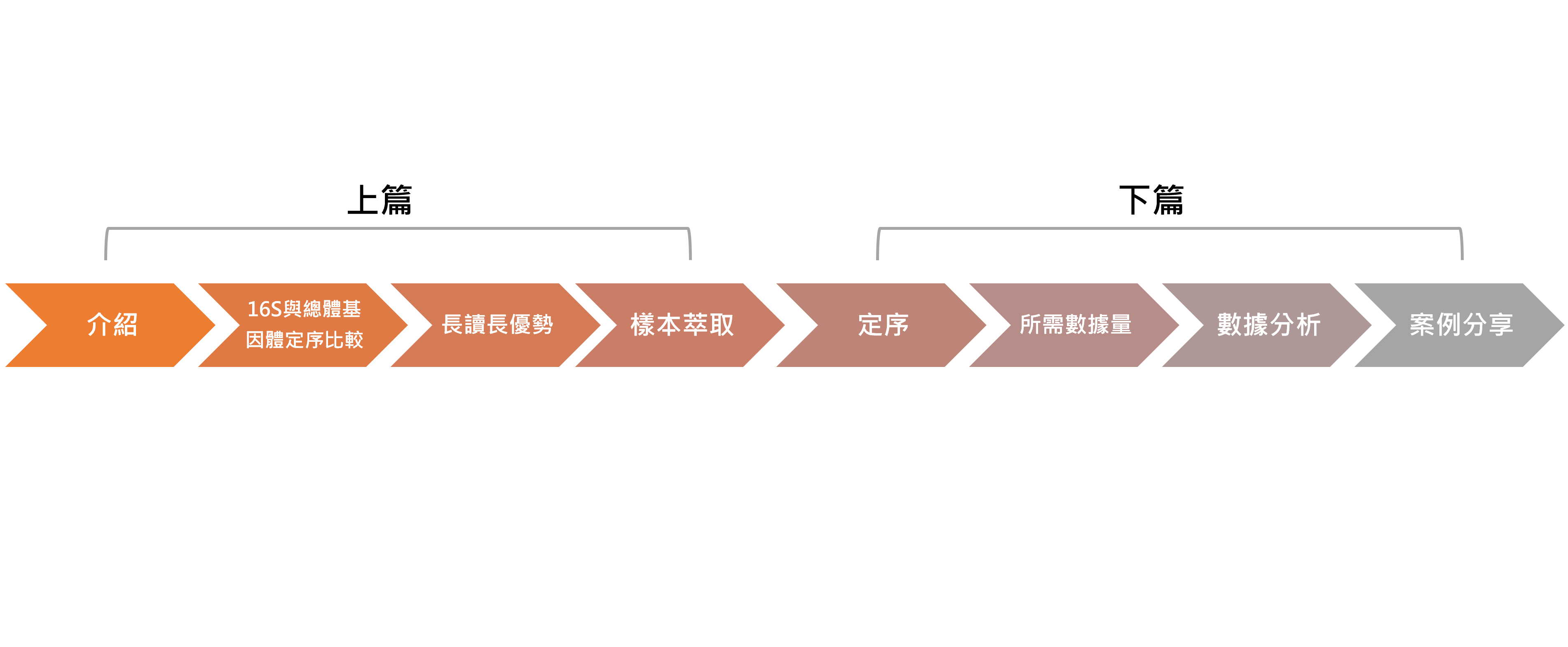
各位,元旦快樂 ~ 接下來就讓我們期待過年的 9 天連假吧!! (超認真的真心話) 繼續 (下) 篇的內容吧! (下) 篇的內容包含 Nanopore 定序平台的介紹、不同應用需要的數據量建議、Nanopore 提供之總體基因體學研究相關分析軟體以及案例分享,內容豐富請盡早服用,避免向隅喔~ 前情提要在這裡--你。不只是你。總體基因體學揭開人體內宇宙 (上)
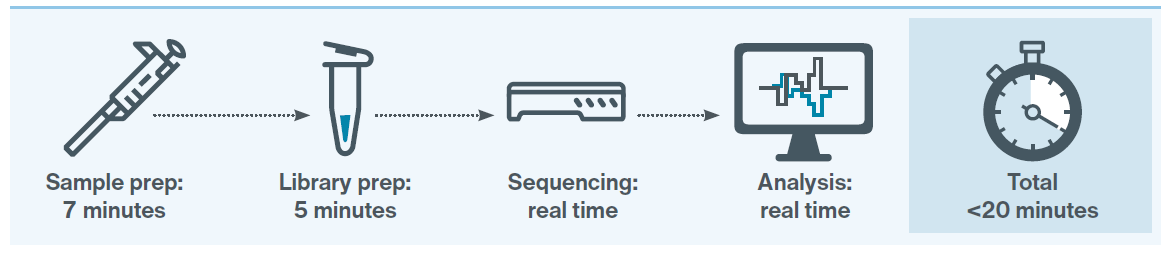
對於許多總體基因體學應用,從檢測到分析結果的總花費時間是至關重要的。例如: 監測疾病爆發或識別危及生命的病原微生物感染等,都需要快速獲得準確的基因體訊息。 Nanopore 定序不同於傳統的定序技術,傳統的定序技術通常在定序結束時批量產出數據,而 Nanopore 定序可即時產生數據用於分析,搭配選擇快速建庫套組,極大的縮短製備時間,整個流程約可在 20 分鐘內完成,因此可以有效且具經濟效益地識別不同的微生物,將檢測的時程縮短在幾分鐘內而不是幾天內消滅病原體(Fig. 1)。
(Fig. 1)
定序平台
Nanopore 除了提供廣為人知的攜帶式定序設備--- MinION 以及 Flongle , 近年也陸續推出了高數據產出量之平台,包含 GridION、 PromethION 24 和 PromethION 48 設備 (Fig. 2),在今年 2021 年底更是於 PromethION 系列增加了 PromethION 2 (P2) 以及 P2 Solo。其中 Flongle 與 MinION 提供了極端環境研究者隨時隨地定序樣本的條件; 而 PromethION 提供最大數據量與最低定序成本。給予不同需求的研究者彈性的選擇。

(Fig. 2)
下列表格比較了不同定序設備之規格以及數據品質:


建議數據量
Nanopore 針對不同的總體基因體研究目的,提供了建議的定序數據量,但須注意其只應作為大約的定序深度判斷,實際的定序需求量取決於實驗目標和樣本,例如: 微生物的稀有性、樣本的複雜性、宿主 DNA 的存在等。
使用全基因體方法定序總體基因體 (Metagenomics),依據不同研究目的的推薦數據覆蓋度如下:
*注意: 以下為單個生物體之覆蓋度
• 確認感興趣的生物體的存在:10x
• 物種級別 ID:20x
• AMR 基因分析:20x
• 組裝:30x
•識別變異:100x
Nanopore 提供之微生物研究相關分析流程
現在有許多工具可使用 Nanopore 數據進行總體基因體分析—從快速物種鑑定到組裝來自混合微生物樣本的完整基因體。 EPI2ME 是 Nanopore 提供的一個基於雲端的數據分析平台,能夠即時對 Nanopore 數據進行端到端的分析,內含多個工作流程 (Fig. 3)。EPI2ME 具使用者介面,直觀的圖形介面有助於解釋單個或多個混樣樣本方便操作者輕鬆運行。分析會顯示完整的 QC 指標項目,提供有關定序性能的反饋,包括 Reads 數、讀長分佈和 Q值分數等。

(Fig. 3)
在微生物相關分析上, EPI2ME 提供了多種量身定制的工作流程,以 WIMP: What’s in my pot? 為基礎,提供完整的抗微生物耐藥性 (AMR) 分析,其在使用上不需要生物資訊學專業知識,使任何研究人員都能夠快速和輕鬆地進行樣品分析。 另外,Nanopore 還提供 EPI2ME 16S 分析工作流程,可快速鑑定屬級 (Genus level) 細菌。16S 基因兼具保守性和高度變異區,其存在於所有細菌和古細菌。
以下表格整理了 EPI2ME 提供之微生物相關分析工具:

組裝工具
在使用 Nanopore 進行模擬微生物群落定序時,Nanopore 產出之一致性序列已超過 Q40 (99.99%) 的準確率,足夠支持後續高度準確的基因體組裝和功能研究。在總體基因體樣本的組裝 Nanopore 建議使用第三方從頭組裝工具 MetaFlye [4],MetaFlye 是一個完整的組裝流程,將原始 Nanopore 序列作為輸入並產生拋光的 Contigs 作為輸出。此外, Nanopore 也推薦使用 Medaka [5] 對完成的組裝再進行一輪額外拋光。這些工具可以在 GitHub 上找到。 在分析時間的部分,對於 30 Gb 的總體基因體數據,用 MetaFlye 組裝需要 18 小時,併用 Medaka 的話需要再加上 2-4 小時(基於 8 個 CPU 的計算);Nanopore 建議至少需要 100 GB RAM 用於執行 MetaFlye。 當然,除了MetaFlye,也可以選擇使用熱門的組裝工具 Canu 和 Flye,Latorre-Perez et al. [6] 描述了使用 Canu 和 Flye 組裝了高度完整且連續了模擬微生物群落的基因體草圖。
案例分享之一---【高品質人類糞便萃取指南與分析流程】[7]

雖然長讀長定序已經成功應用於連續細菌分離株的基因體組裝。儘管如此,從糞便中萃取高度完整性、足夠總量、純度的 DNA 用於總基因體定序仍然是一項長期研究障礙。本篇分享之研究團隊提出可從人類糞便檢體萃取足量 HMW DNA的方法以及用於下游數據分析的分析流程: Lathe (https://github.com/bhattlab/lathe)。 研究團隊提出之萃取protocol極致詳盡,包含多個酵素裂解及片段篩選步驟。可由總量300-500mg的樣品中萃取1-2 µg的HMW DNA,片段範圍坐落於15-50 kb,於MinION R9.4 上可生成約 6-30 Gbp 的數據。除了詳列所需試劑與萃取步驟,本篇也貼心列出可能遇到的狀況與解決方法,總萃取時間可在8小時完成。 另外,由於長讀長的準確率不若NGS,研究團隊還還開發了一個用於鹼基識別、易錯序列組裝和環狀化的下游分析工作流程: Lathe。鹼基識別選擇 Guppy basecaller; 組裝可依照需求選用Canu 或 Flye; 組裝完成後長讀長拋光使用Racon及Medaka; 若使用短讀長拋光則使用Pilon,在大多數情況下,使用短讀長拋光是首選,因為它提高了準確度和速度,然而,當短讀長覆蓋不均勻的情況下,建議可以透過加入長讀長拋光步驟來改進。本篇研究指南僅需2天的手動實驗與計算,總共可在10天內完成從複雜的人類腸道樣本中產生高品質的連續或環狀細菌基因體。
案例分享之二---【快速病原體及AMR分析】[8]

本篇分享之團隊使用可提供近乎實時 DNA 序列分析的 MinION 定序平台以及團隊自行開發之 NanoOK RT 工具進行散彈槍總體基因體定序並分析 mock community 和健康和壞死性小腸結腸炎(Necrotising enterocolitis ;NEC)早產兒的糞便樣本,以評估其腸道相關微生物菌相之差異。
據 PCoA 結果顯示 NEC 和健康早產嬰兒菌相組成具有顯著差異,其中,罹患 NEC 早產兒組別內含之主要致病菌 Klebsiella pneumoniae 及 Enterobacter cloacae 及其相應的 AMR 基因圖譜在短短 1 小時的定序時間內即可偵測到。其結果通過病原體分離、全基因體定序和抗生素藥物敏感試驗,以及具有已知 AMR 基因的 mock community 和臨床樣本得到驗證。 研究團隊表示,結合 NanoOK RT 與 MinION(或是具有成本效益的 Flongle )可以在 < 5 小時內將總體基因體樣本轉換為分析完成之數據結果,為未來的研究創造了一個平台,以期在臨床環境中開發這些工具和方法,提供量身定制的患者抗菌治療方案。
在過去,微生物研究的進展受到許多因素限制,包括絕大多數無法在人工培養基上培養的生物。然而,隨著定序技術的進步,我們開始能夠真正了解微生物的多樣性和相互作用。雖然目前仍然存在許多挑戰,例如基因體組裝、定序周轉時間和定序基礎設施要求等。 現在,Oxford Nanopore 正在解決這些挑戰,讓研究人員可以在實驗室和採樣點進行完整檢測,改變目前從採樣到數據結果交付需要數月的時程,將其縮短至數小時即可完成現場工作,以允許快速和果斷的應對行動。
參考資料
1. Oxford Nanopore Technologies. What’s in my Pot? (WIMP), a quantitative analysis tool for realtime species identification. Poster. Available at: https://nanoporetech.com/resource-centre/whats-mypot-wimp-quantitative-analysistool-real-time-species-identification [Accessed: 18 February 2020]
2. Oxford Nanopore Technologies. Real-time detection of antibioticresistance genes using Oxford Nanopore Technologies’ MinION. Poster. Available at: https://nanoporetech.com/resource-centre/real-time-detection-antibioticresistance-genes-using-oxfordnanopore-technologies [Accessed:18 February 2020]
3. Oxford Nanopore Technologies. Barcode of life: simple laboratory workflows for 16S and CO1 genus and species identification. Poster. Available at: https://nanoporetech.com/resource-centre/barcodelife-simple-laboratory-workflows-16s-and-co1-genus-and-species.
4. Kolmogorov, M. et al. MetaFlye: scalable long-read metagenome assembly using repeat graphs. Nat Methods. 17: 1103-1110 (2020).
5. Oxford Nanopore Technologies. Medaka (v1.4.3). Software available at: github.com/nanoporetech/medaka.
6. Nicholls, S.M, Quick, J.C., Tang, S., and Loman, N.J. Ultra-deep, longread nanopore sequencing of mock microbial community standards. GigaScience. 8(5) giz043 (2019).
7. Maghini, D.G., Moss, E.L., Vance, S.E. et al. Improved high-molecular-weight DNA extraction, nanopore sequencing and metagenomic assembly from the human gut microbiome. Nat Protoc 16, 458–471 (2021). https://doi.org/10.1038/s41596-020-00424-x
8. Leggett, R.M., Alcon-Giner, C., Heavens, D. et al. Rapid MinION profiling of preterm microbiota and antimicrobial-resistant pathogens. Nat Microbiol 5, 430–442 (2020). https://doi.org/10.1038/s41564-019-0626-z
9. Oxford Nanopore Technologies. Whole genome sequencing and assembly white paper. https://nanoporetech.com/resource-centre/whole-genome-sequencing-and-assembly-white-paper
10. Oxford Nanopore Technologies. Workflow: Metagenomic assembly. https://nanoporetech.com/resource-centre/literature/metagenomic-assembly-workflow
11. Oxford Nanopore Technologies. metagenomics-whitepaper. https://nanoporetech.com/white-papers/metagenomics

各位,元旦快樂 ~ 接下來就讓我們
對於許多總體基因體學應用,從檢測到分析結果的總花費時間是至關重要的。例如: 監測疾病爆發或識別危及生命的病原微生物感染等,都需要快速獲得準確的基因體訊息。 Nanopore 定序不同於傳統的定序技術,傳統的定序技術通常在定序結束時批量產出數據,而 Nanopore 定序可即時產生數據用於分析,搭配選擇快速建庫套組,極大的縮短製備時間,整個流程約可在 20 分鐘內完成,因此可以有效且具經濟效益地識別不同的微生物,將檢測的時程縮短在幾分鐘內而不是幾天內消滅病原體(Fig. 1)。

(Fig. 1)
定序平台
Nanopore 除了提供廣為人知的攜帶式定序設備--- MinION 以及 Flongle , 近年也陸續推出了高數據產出量之平台,包含 GridION、 PromethION 24 和 PromethION 48 設備 (Fig. 2),在今年 2021 年底更是於 PromethION 系列增加了 PromethION 2 (P2) 以及 P2 Solo。其中 Flongle 與 MinION 提供了極端環境研究者隨時隨地定序樣本的條件; 而 PromethION 提供最大數據量與最低定序成本。給予不同需求的研究者彈性的選擇。

(Fig. 2)
下列表格比較了不同定序設備之規格以及數據品質:


建議數據量
Nanopore 針對不同的總體基因體研究目的,提供了建議的定序數據量,但須注意其只應作為大約的定序深度判斷,實際的定序需求量取決於實驗目標和樣本,例如: 微生物的稀有性、樣本的複雜性、宿主 DNA 的存在等。
使用全基因體方法定序總體基因體 (Metagenomics),依據不同研究目的的推薦數據覆蓋度如下:
*注意: 以下為單個生物體之覆蓋度
• 確認感興趣的生物體的存在:10x
• 物種級別 ID:20x
• AMR 基因分析:20x
• 組裝:30x
•識別變異:100x
Nanopore 提供之微生物研究相關分析流程
現在有許多工具可使用 Nanopore 數據進行總體基因體分析—從快速物種鑑定到組裝來自混合微生物樣本的完整基因體。 EPI2ME 是 Nanopore 提供的一個基於雲端的數據分析平台,能夠即時對 Nanopore 數據進行端到端的分析,內含多個工作流程 (Fig. 3)。EPI2ME 具使用者介面,直觀的圖形介面有助於解釋單個或多個混樣樣本方便操作者輕鬆運行。分析會顯示完整的 QC 指標項目,提供有關定序性能的反饋,包括 Reads 數、讀長分佈和 Q值分數等。

(Fig. 3)
在微生物相關分析上, EPI2ME 提供了多種量身定制的工作流程,以 WIMP: What’s in my pot? 為基礎,提供完整的抗微生物耐藥性 (AMR) 分析,其在使用上不需要生物資訊學專業知識,使任何研究人員都能夠快速和輕鬆地進行樣品分析。 另外,Nanopore 還提供 EPI2ME 16S 分析工作流程,可快速鑑定屬級 (Genus level) 細菌。16S 基因兼具保守性和高度變異區,其存在於所有細菌和古細菌。
以下表格整理了 EPI2ME 提供之微生物相關分析工具:

組裝工具
在使用 Nanopore 進行模擬微生物群落定序時,Nanopore 產出之一致性序列已超過 Q40 (99.99%) 的準確率,足夠支持後續高度準確的基因體組裝和功能研究。在總體基因體樣本的組裝 Nanopore 建議使用第三方從頭組裝工具 MetaFlye [4],MetaFlye 是一個完整的組裝流程,將原始 Nanopore 序列作為輸入並產生拋光的 Contigs 作為輸出。此外, Nanopore 也推薦使用 Medaka [5] 對完成的組裝再進行一輪額外拋光。這些工具可以在 GitHub 上找到。 在分析時間的部分,對於 30 Gb 的總體基因體數據,用 MetaFlye 組裝需要 18 小時,併用 Medaka 的話需要再加上 2-4 小時(基於 8 個 CPU 的計算);Nanopore 建議至少需要 100 GB RAM 用於執行 MetaFlye。 當然,除了MetaFlye,也可以選擇使用熱門的組裝工具 Canu 和 Flye,Latorre-Perez et al. [6] 描述了使用 Canu 和 Flye 組裝了高度完整且連續了模擬微生物群落的基因體草圖。
案例分享之一---【高品質人類糞便萃取指南與分析流程】[7]

雖然長讀長定序已經成功應用於連續細菌分離株的基因體組裝。儘管如此,從糞便中萃取高度完整性、足夠總量、純度的 DNA 用於總基因體定序仍然是一項長期研究障礙。本篇分享之研究團隊提出可從人類糞便檢體萃取足量 HMW DNA的方法以及用於下游數據分析的分析流程: Lathe (https://github.com/bhattlab/lathe)。 研究團隊提出之萃取protocol極致詳盡,包含多個酵素裂解及片段篩選步驟。可由總量300-500mg的樣品中萃取1-2 µg的HMW DNA,片段範圍坐落於15-50 kb,於MinION R9.4 上可生成約 6-30 Gbp 的數據。除了詳列所需試劑與萃取步驟,本篇也貼心列出可能遇到的狀況與解決方法,總萃取時間可在8小時完成。 另外,由於長讀長的準確率不若NGS,研究團隊還還開發了一個用於鹼基識別、易錯序列組裝和環狀化的下游分析工作流程: Lathe。鹼基識別選擇 Guppy basecaller; 組裝可依照需求選用Canu 或 Flye; 組裝完成後長讀長拋光使用Racon及Medaka; 若使用短讀長拋光則使用Pilon,在大多數情況下,使用短讀長拋光是首選,因為它提高了準確度和速度,然而,當短讀長覆蓋不均勻的情況下,建議可以透過加入長讀長拋光步驟來改進。本篇研究指南僅需2天的手動實驗與計算,總共可在10天內完成從複雜的人類腸道樣本中產生高品質的連續或環狀細菌基因體。
案例分享之二---【快速病原體及AMR分析】[8]

本篇分享之團隊使用可提供近乎實時 DNA 序列分析的 MinION 定序平台以及團隊自行開發之 NanoOK RT 工具進行散彈槍總體基因體定序並分析 mock community 和健康和壞死性小腸結腸炎(Necrotising enterocolitis ;NEC)早產兒的糞便樣本,以評估其腸道相關微生物菌相之差異。
據 PCoA 結果顯示 NEC 和健康早產嬰兒菌相組成具有顯著差異,其中,罹患 NEC 早產兒組別內含之主要致病菌 Klebsiella pneumoniae 及 Enterobacter cloacae 及其相應的 AMR 基因圖譜在短短 1 小時的定序時間內即可偵測到。其結果通過病原體分離、全基因體定序和抗生素藥物敏感試驗,以及具有已知 AMR 基因的 mock community 和臨床樣本得到驗證。 研究團隊表示,結合 NanoOK RT 與 MinION(或是具有成本效益的 Flongle )可以在 < 5 小時內將總體基因體樣本轉換為分析完成之數據結果,為未來的研究創造了一個平台,以期在臨床環境中開發這些工具和方法,提供量身定制的患者抗菌治療方案。
在過去,微生物研究的進展受到許多因素限制,包括絕大多數無法在人工培養基上培養的生物。然而,隨著定序技術的進步,我們開始能夠真正了解微生物的多樣性和相互作用。雖然目前仍然存在許多挑戰,例如基因體組裝、定序周轉時間和定序基礎設施要求等。 現在,Oxford Nanopore 正在解決這些挑戰,讓研究人員可以在實驗室和採樣點進行完整檢測,改變目前從採樣到數據結果交付需要數月的時程,將其縮短至數小時即可完成現場工作,以允許快速和果斷的應對行動。
參考資料
1. Oxford Nanopore Technologies. What’s in my Pot? (WIMP), a quantitative analysis tool for realtime species identification. Poster. Available at: https://nanoporetech.com/resource-centre/whats-mypot-wimp-quantitative-analysistool-real-time-species-identification [Accessed: 18 February 2020]
2. Oxford Nanopore Technologies. Real-time detection of antibioticresistance genes using Oxford Nanopore Technologies’ MinION. Poster. Available at: https://nanoporetech.com/resource-centre/real-time-detection-antibioticresistance-genes-using-oxfordnanopore-technologies [Accessed:18 February 2020]
3. Oxford Nanopore Technologies. Barcode of life: simple laboratory workflows for 16S and CO1 genus and species identification. Poster. Available at: https://nanoporetech.com/resource-centre/barcodelife-simple-laboratory-workflows-16s-and-co1-genus-and-species.
4. Kolmogorov, M. et al. MetaFlye: scalable long-read metagenome assembly using repeat graphs. Nat Methods. 17: 1103-1110 (2020).
5. Oxford Nanopore Technologies. Medaka (v1.4.3). Software available at: github.com/nanoporetech/medaka.
6. Nicholls, S.M, Quick, J.C., Tang, S., and Loman, N.J. Ultra-deep, longread nanopore sequencing of mock microbial community standards. GigaScience. 8(5) giz043 (2019).
7. Maghini, D.G., Moss, E.L., Vance, S.E. et al. Improved high-molecular-weight DNA extraction, nanopore sequencing and metagenomic assembly from the human gut microbiome. Nat Protoc 16, 458–471 (2021). https://doi.org/10.1038/s41596-020-00424-x
8. Leggett, R.M., Alcon-Giner, C., Heavens, D. et al. Rapid MinION profiling of preterm microbiota and antimicrobial-resistant pathogens. Nat Microbiol 5, 430–442 (2020). https://doi.org/10.1038/s41564-019-0626-z
9. Oxford Nanopore Technologies. Whole genome sequencing and assembly white paper. https://nanoporetech.com/resource-centre/whole-genome-sequencing-and-assembly-white-paper
10. Oxford Nanopore Technologies. Workflow: Metagenomic assembly. https://nanoporetech.com/resource-centre/literature/metagenomic-assembly-workflow
11. Oxford Nanopore Technologies. metagenomics-whitepaper. https://nanoporetech.com/white-papers/metagenomics
圖爾思生物科技 / 微生物體研究中心
吳雁韻 文案



