09
2023.06
ddRAD-seq 定序古拳法 -人人皆能成為成本效益大師
ddRAD (Double Digest Restriction Associated DNA) - 從案件評估、建庫、定序到分子標誌。
對於進行遺傳多樣性研究、 QTL mapping、GWAS 等等的研究人員而言,在取得合適的研究族群後,所面臨的問題將是如何大規模且有效的取得變異位點。
在定序價格持續下降的現在,以全基因體定序 (Whole genome sequencing, WGS) 定序整個物種基因體不再昂貴,但對於樣品數動輒上百的遺傳研究仍是一筆可觀的費用,同時 WGS 技術產出的大量序列,在分析和數據儲存上可能會耗費許多不必要的資源。因此簡化基因體定序 (Reduced-representation sequencing, RRS) 仍然有經濟和快速的優勢。
簡化基因體定序技術包含 RRL (Reduced-Representation Library)、RADseq (Restriction-site Associated DNA sequencing)、GBS (Genotyping by sequencing) 等不同建庫方法,可適用於無參考序列的物種。建庫後僅定序特定切位旁固定長度的片段,被定序的部分常為該物種全基因體的 5% 或是更少。
圖爾思提供 ddRAD (Double Digest Restriction Associated DNA) 建庫方法 (Peterson et al., 2012),使用兩個限制酶 (Restriction Enzyme) 進行酶切,並搭配片段篩選 (Size selection) 進行基因體簡化,可以穩定控制定序片段區域,且可根據需求更換限制酶組合,給予建庫流程更多彈性(圖一)。
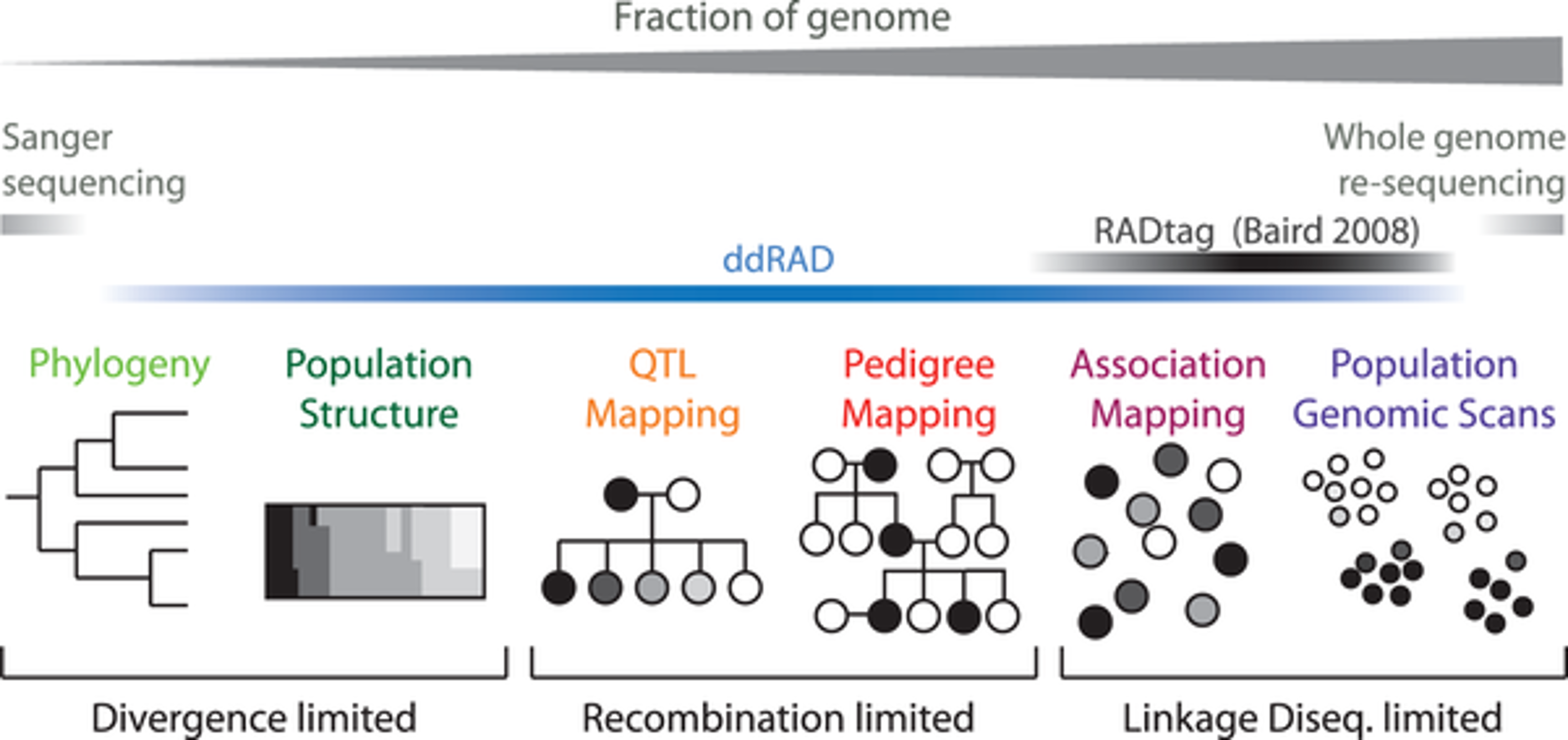
圖一、不同應用所需分子標誌數量與 genotyping 策略 (Peterson et al., 2012)
在案件評估階段,針對有參考序列的物種,可使用圖爾思自建 Biotools ddRAD 雲平台進行序列切位模擬,目前平台提供 11 個物種參考序列和 106 組常用限制酶模擬(圖二)。
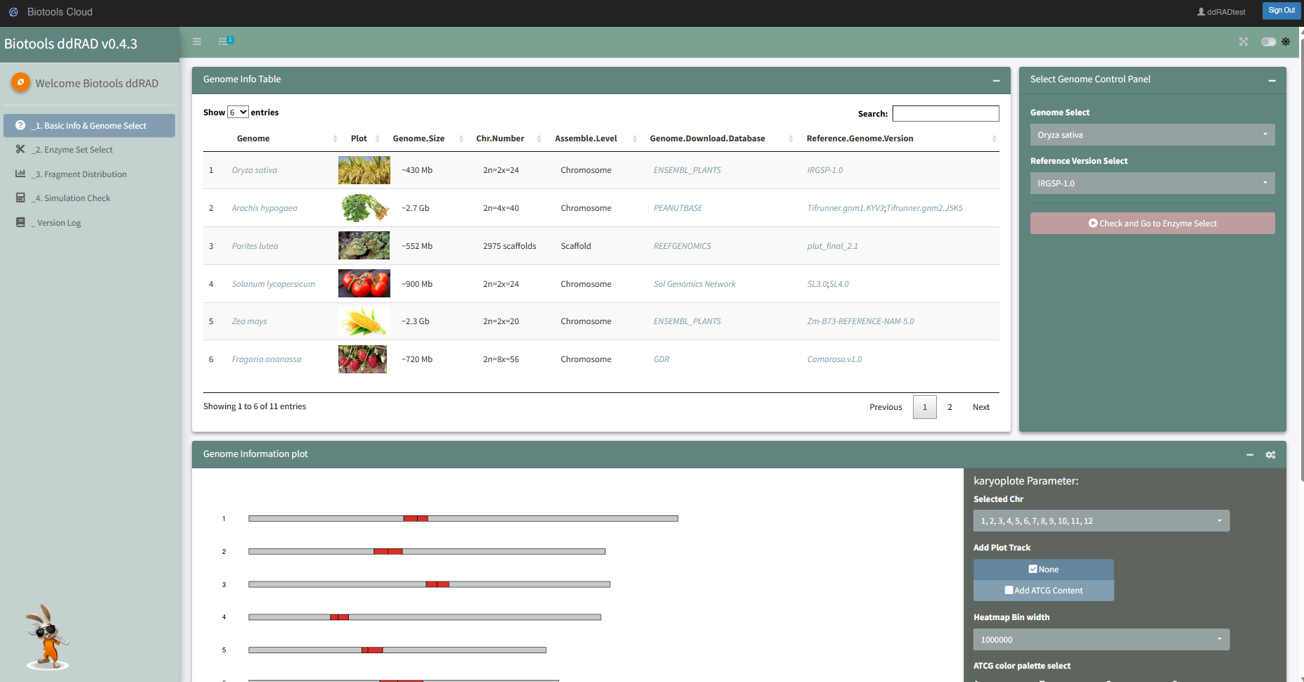
圖二、Biotools ddRAD 雲平台介面
in silico 切位模擬介面可以選擇限制酶 Rare Cutter (Enzyme A)、Common Cutter (Enzyme B)、以及 size seletion 範圍,即可得到以參考序列模擬,以兩端切位命名的 AA、AB 和 BB 片段,評估實驗條件的重點如下:
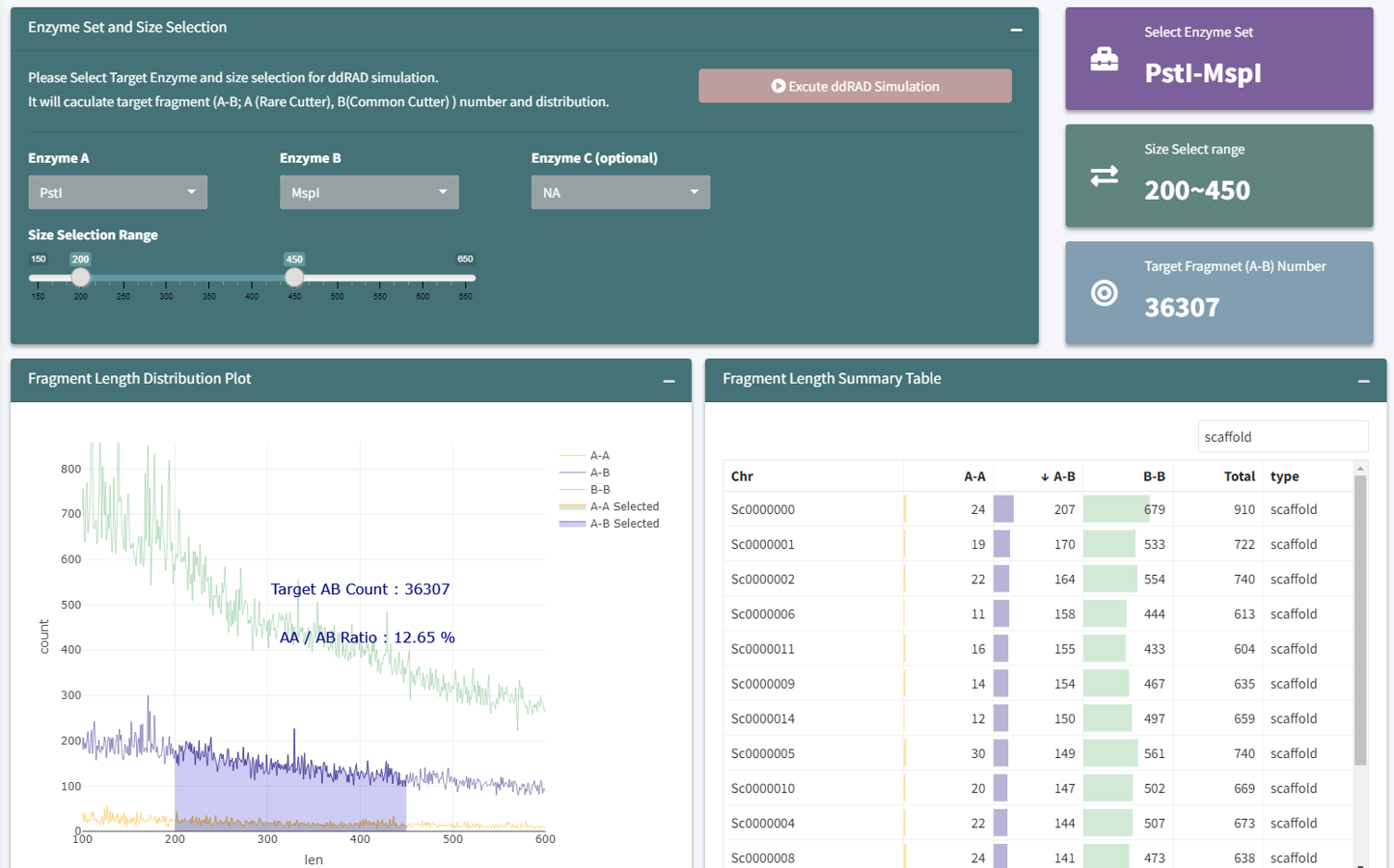
圖三、Biotools ddRAD 雲平台切位模擬介面
表一、限制酶組合模擬結果
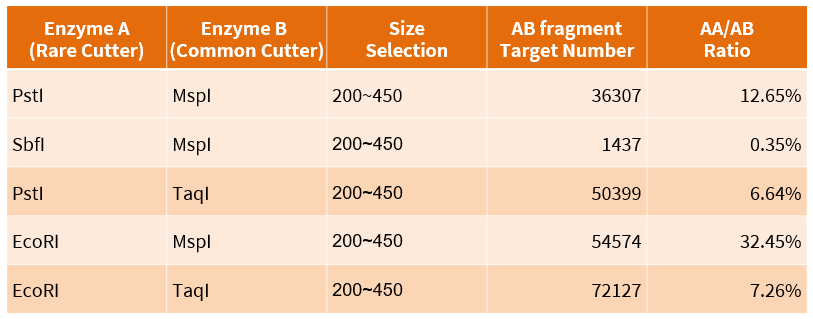
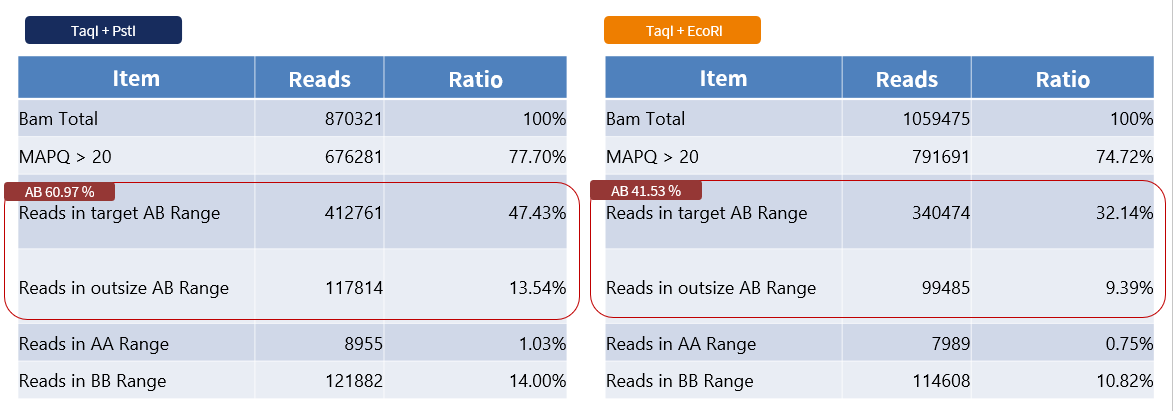
前測試選擇兩個親緣關係較遠的個體,以 TaqI+PstI 和 TaqI+EcoRI 限制酶組合進行建庫,分別得到 13,739 和 12,431 個同質多型性位點。
由於該物種的參考序列與實際樣品親緣較遠,且相對較不完整,模擬的正確度可能會受到影響,因此將實際定序序列比對回模擬結果,發現 TaqI+PstI 這個限制酶組合較符合模擬結果。
表二、實際序列比對模擬結果
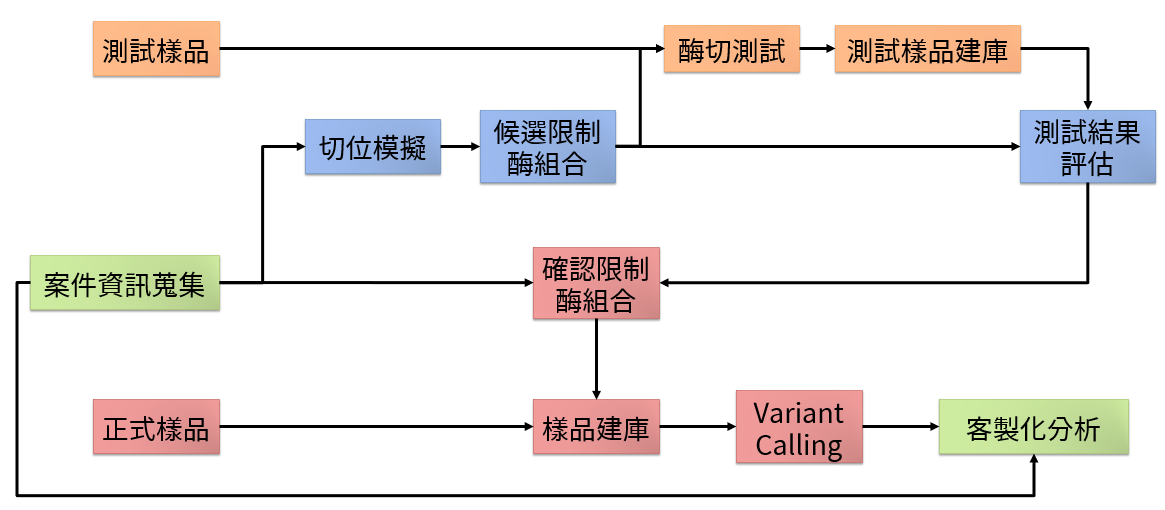
圖三、ddRAD 案件流程
回上一頁
對於進行遺傳多樣性研究、 QTL mapping、GWAS 等等的研究人員而言,在取得合適的研究族群後,所面臨的問題將是如何大規模且有效的取得變異位點。
在定序價格持續下降的現在,以全基因體定序 (Whole genome sequencing, WGS) 定序整個物種基因體不再昂貴,但對於樣品數動輒上百的遺傳研究仍是一筆可觀的費用,同時 WGS 技術產出的大量序列,在分析和數據儲存上可能會耗費許多不必要的資源。因此簡化基因體定序 (Reduced-representation sequencing, RRS) 仍然有經濟和快速的優勢。
簡化基因體定序技術包含 RRL (Reduced-Representation Library)、RADseq (Restriction-site Associated DNA sequencing)、GBS (Genotyping by sequencing) 等不同建庫方法,可適用於無參考序列的物種。建庫後僅定序特定切位旁固定長度的片段,被定序的部分常為該物種全基因體的 5% 或是更少。
圖爾思提供 ddRAD (Double Digest Restriction Associated DNA) 建庫方法 (Peterson et al., 2012),使用兩個限制酶 (Restriction Enzyme) 進行酶切,並搭配片段篩選 (Size selection) 進行基因體簡化,可以穩定控制定序片段區域,且可根據需求更換限制酶組合,給予建庫流程更多彈性(圖一)。

圖一、不同應用所需分子標誌數量與 genotyping 策略 (Peterson et al., 2012)
在案件評估階段,針對有參考序列的物種,可使用圖爾思自建 Biotools ddRAD 雲平台進行序列切位模擬,目前平台提供 11 個物種參考序列和 106 組常用限制酶模擬(圖二)。

圖二、Biotools ddRAD 雲平台介面
- AB 片段數量 -> AB 片段為定序目標,可依據族群結構和後續應用調整所需片段數量
- AA/AB ratio -> 越低越好,AA 序列不會被定序,但會影響建庫定量結果
- 限制酶價格 -> 選用單價較低的限制酶可節省建庫成本

圖三、Biotools ddRAD 雲平台切位模擬介面
實際案例分享:
以下是某物種參考過去文獻所挑出的數個限制酶組合,以雲平台所做出的切位模擬結果。
考量 AB 片段數量以及 AA/AB ratio,最終選擇 TaqI+PstI 和 TaqI+EcoRI 進行前測試。
考量 AB 片段數量以及 AA/AB ratio,最終選擇 TaqI+PstI 和 TaqI+EcoRI 進行前測試。
表一、限制酶組合模擬結果

前測試選擇兩個親緣關係較遠的個體,以 TaqI+PstI 和 TaqI+EcoRI 限制酶組合進行建庫,分別得到 13,739 和 12,431 個同質多型性位點。
由於該物種的參考序列與實際樣品親緣較遠,且相對較不完整,模擬的正確度可能會受到影響,因此將實際定序序列比對回模擬結果,發現 TaqI+PstI 這個限制酶組合較符合模擬結果。
表二、實際序列比對模擬結果

案件流程:
- ddRAD 案件屬特殊案件,請先與專員討論實驗設計以及相關資訊。
- 若實驗物種有參考序列,可評估是否可進行切位模擬。
- 首次進行 ddRAD 建庫,建議先以少量樣品進行前測試。
- 除 Variant calling 之外的分析皆屬客製化分析,有相關需求請與專員詳談。

圖三、ddRAD 案件流程
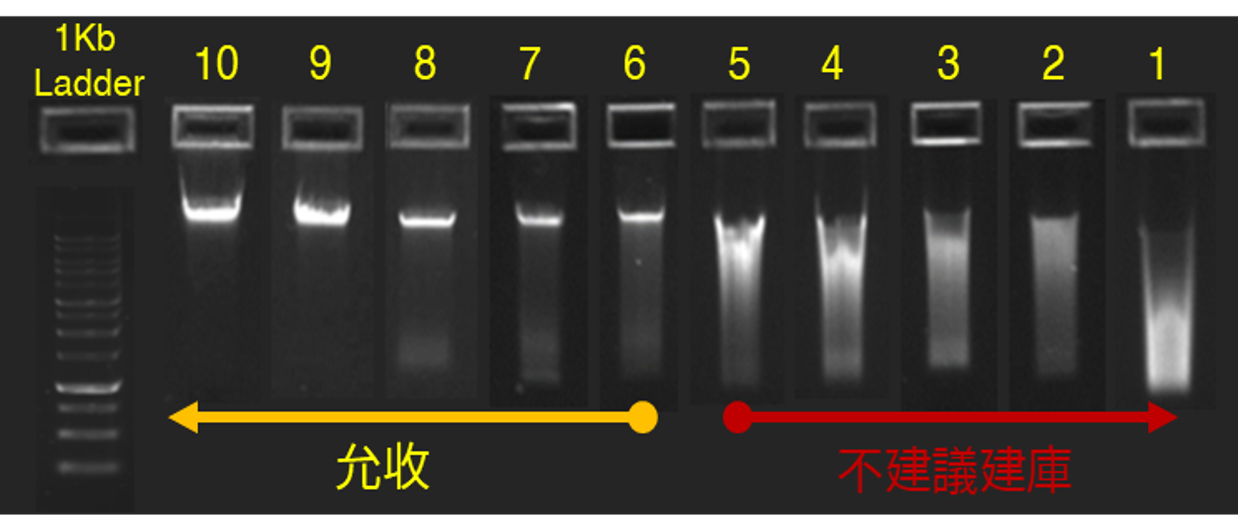
樣品收件標準:
- High Molecular Weight Genomic DNA, HMW DNA ≧ 1000 ng
- DNA 濃度 ≧ 20 ng/ ul (Qubit測定)
- 樣品體積 ≧ 50 ul
- 樣本必須要有完整 Genome片段(主片段,如下圖 6-10號樣本)
建議主要DNA片段 ≧ 10 kb - OD260/OD280 = 1.8-2.0
- OD260/OD230 ≧ 2.0

參考資料:
B.K. Peterson, et al., Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species, 2012/05/31 edn, PLoS One 7 (5) (2012)



